Conduct disorder and its related disruptive behavior disorders are among the most common forms of psychopathology in children and adolescents. In children, severe disruptive behaviors interfere with interpersonal relationships and academic success. If left untreated, these disorders can lead to additional problems during the critical developmental years and later in life, including juvenile delinquency, substance abuse, and criminal activity
(1–
3). The treatment of persistent, severe behavioral problems necessitates a multidisciplinary approach, including psychosocial and psychopharmacological interventions.
Antipsychotics are commonly used in children to target aggression and other problem behaviors related to disruptive behavior disorders and other neuropsychiatric conditions
(4). The atypical antipsychotic most frequently studied in this clinical setting is risperidone
(5–
17). In studies of children with disruptive behavior disorders who also had developmental disabilities, risperidone was associated with an improvement in target behaviors related to disruptive behavior disorders
(6–
8), nonspecific behavior problems or self-injurious behavior
(9,
10), and pervasive developmental disorders
(11–
17).
Several challenges are unique to studying children with developmental disabilities and comorbid neuropsychiatric diagnoses. Difficulty in obtaining adequate numbers of patients of this type, problems related to working with patients with poor cognitive and verbal skills, and difficulty obtaining informed consent have resulted in a dearth of large, well-controlled, long-term studies in this population
(18). Because many disorders associated with severe behavioral abnormalities are chronic, long-term treatment is usually needed. In a study by Turgay et al.
(8), ongoing symptom reduction, moderate weight gain, and an initial rise in prolactin concentrations were observed in 77 children with subaverage intelligence who were treated with risperidone over 48 weeks. The study reported here is a larger-scale, long-term, prospective trial of risperidone in children with developmental disabilities and disruptive behavior disorders.
Method
This 48-week, open-label, multicenter, follow-up study was designed to examine the long-term safety and effectiveness of risperidone in children previously enrolled in a 6-week, multicenter, double-blind, placebo-controlled trial of risperidone
(19). In that study of 118 disruptive children ages 5–12 years with subaverage intelligence (IQ ≥36 and ≤84, inclusive), the patients received 0.02 to 0.06 mg/kg/day of risperidone oral solution or placebo. The primary efficacy measure was the change from baseline to endpoint on the conduct problem subscale of the Nisonger Child Behavior Rating Form
(20).
To be eligible for the extension study, all patients had to have completed at least 2 weeks of the double-blind trial. Patients who discontinued the double-blind trial after 2 weeks but before study completion for reasons other than adverse events were eligible to enter the extension phase. The patients were required to continue to meet many of the inclusion criteria of the double-blind study, as follows:
1.
Being ages 5–12 years when enrolled in the preceding 6-week study
2.
Having a primary DSM-IV axis I diagnosis of conduct disorder, oppositional defiant disorder, or disruptive behavior disorder not otherwise specified
3.
Having a DSM-IV axis II diagnosis of borderline intellectual functioning or mild to moderate mental retardation (IQ 36–84, as measured on the Stanford-Binet or Wechsler Intelligence Scales)
4.
Having a total rating ≥24 on the conduct problem subscale of the Nisonger Child Behavior Rating Form
5.
Having behavioral symptoms before treatment that were sufficiently severe that further antipsychotic therapy was deemed necessary by an investigator
Written informed consent was provided by the participants (if capable) and their guardian or legal representative before they entered the extension trial.
Exclusion criteria included experiencing significant adverse events possibly attributed to risperidone during the double-blind study; having any adverse event resulting in discontinuation from the double-blind study; completing the double-blind study more than 3 weeks ago; having extrapyramidal symptoms that were not adequately controlled; having a diagnosis of pervasive developmental disorder, schizophrenia, or other psychotic disorder; having a seizure disorder requiring medication; and the use of disallowed medications, to be described.
Study Design
Patient demographic and clinical characteristics were recorded as part of the double-blind trial. For the patients who entered the study within 10 days of completing the double-blind trial, the final safety and efficacy assessments made during the earlier trial served as baseline data for the extension study. The patients who entered the study after 10 days had elapsed were reassessed.
Medications
Study treatment from the double-blind trial (oral risperidone, 0.006–0.092 mg/kg/day or placebo) was discontinued at completion of the double-blind study or withdrawal, after which all enrolled patients received open-label risperidone. On days 1 and 2, the patients received 0.01 mg/kg of risperidone oral solution. On day 3, the dose was increased to 0.02 mg/kg. Thereafter, the dose could be further adjusted at weekly intervals, up to a maximum allowable dose of 0.06 mg/kg/day. The entire dose was administered in the morning unless breakthrough symptoms were noted in the afternoon or evening, in which case, risperidone could be given twice daily.
Psychostimulant medications used in a constant dose throughout the study were permitted for patients already taking these drugs for at least 30 days for attention deficit hyperactivity disorder. Antihistamines, chloral hydrate, and melatonin to promote sleep were permitted under restricted conditions. Anticholinergic medications commonly used to treat extrapyramidal symptoms (e.g., biperiden, trihexyphenidyl, benztropine, procyclidine, and diphenhydramine) were permitted if extrapyramidal symptoms emerged during the study and did not respond to a lower risperidone dose. No other concomitant medications were permitted.
Safety Assessments
Study visits were scheduled at the baseline of the extension study (day 1), days 7, 14, 21, and 28, and every 4 weeks from weeks 8 through 48. At every visit, the investigator recorded each participant’s vital signs and any adverse events elicited during an open-ended inquiry. Body weight and clinical laboratory assessments (a urinalysis and standard hematology and biochemistry tests, including a CBC, measures of electrolyte levels, and liver and kidney function tests), as well as growth hormone and prolactin levels, were obtained at baseline and at weeks 4, 12, 24, 36, and 48 before the morning dose. Blood for laboratory assessments was drawn in the morning after an overnight fast and before administration of the morning risperidone dose.
Resting ECGs were obtained at baseline and at weeks 24 and 48, and all were interpreted by the same pediatric cardiologist. Scores on the Extrapyramidal Symptom Rating Scale
(21) were recorded at every visit and before the administration of any antiparkinsonian medication. Sedation was assessed at baseline and at weeks 1, 4, 12, 24, 36, and 48 by using a 100-mm visual analog scale, a commonly used means of evaluating patient-reported clinical phenomena. In using the visual analog scale, a respondent rates the presence of a symptom based on a visual scale on which 0 indicates no symptoms and 100 indicates the worst imaginable intensity.
Cognitive function was assessed at baseline and at weeks 24 and 48 by using a verbal learning task and a two-version continuous performance task. In the easier version of the continuous performance task, the stimuli were displayed on a computer monitor at regular intervals; the more difficult version used short stimulus presentation times with variable intervals between the stimuli, making their appearance less predictable. A patient who made few errors on the easy test (signifying a floor effect) was given the difficult version. The dependent variables included failure to detect the target picture, incorrect responses to the nontarget picture, a total number of correct detections, and a mean response time.
The verbal learning test was a modification of the California Verbal Learning Test, Children’s Version
(22), in which the list of words each child was required to learn comprised 10 of the most basic nouns from the Peabody Picture Vocabulary Test. The examiner read the words at a rate of one per second, and the patients were asked to recall as many as possible (short-delay free recall). After a 10-minute delay, each child was asked to recall as many words as possible (long-delay free recall). Finally, the examiner read a list of 20 words, and each child was asked to indicate whether each word was part of the original list (recognition memory).
Effectiveness Assessments
Scores on the Nisonger Child Behavior Rating Form, the visual analog scale for the most troublesome symptom, and the Clinical Global Impression’s (CGI) disease severity index were obtained at baseline (day 1) and at weeks 4, 12, 24, 36, and 48. The Nisonger Child Behavior Rating Form is a rating scale for assessing behaviors in children with developmental disabilities. The parent version of the Nisonger Child Behavior Rating Form has 10 social competence items divided into two social competence subscales (compliant/calm and adaptive/social) and 60 problem behavior items divided into six problem behavior subscales (conduct problem, insecure or anxious, hyperactive, self-injury or stereotyped, self-isolated or ritualistic, and overly sensitive). Each of the conduct problem items is scored on a 4-point scale (0=behavior did not occur or was not a problem to 3=occurred a lot or was a severe problem). The Nisonger Child Behavior Rating Form has good interrater reliability and good concurrent validity with the widely used Aberrant Behavior Checklist
(23). Because most of the items on the conduct problem subscale of the Nisonger Child Behavior Rating Form correspond to DSM-IV symptoms of disruptive behavior disorders, the change in conduct problem subscale score on the Nisonger Child Behavior Rating Form from the beginning of the extension study to endpoint was chosen as the primary effectiveness measure.
Secondary effectiveness measures were changes from baseline in scores on other Nisonger Child Behavior Rating Form subscales, the visual analog scale for the “most troublesome symptom,” and the investigator’s CGI for severity, as well as the CGI change scores, which compared endpoint with baseline.
Statistical Analysis
Descriptive statistics were provided for demographic variables and baseline characteristics. All patients who took at least one dose of study medication were included in the safety analysis; patients for whom any data were available after open-label initiation of study drug were also included in the effectiveness analysis (the intent-to-treat population). Data were analyzed for patients who received placebo during the double-blind study, those who received risperidone during the double-blind study, and the study cohort as a whole. All statistical tests were interpreted at the 5% significance level (two-tailed).
Adverse events were tabulated by type and incidence. The frequency of abnormalities was determined for clinical laboratory results and for prolactin levels; within-group prolactin level changes from baseline were calculated, and the difference was tested by using two-tailed paired t tests by sex for each treatment.
Descriptive statistics were calculated for scores on the Extrapyramidal Symptom Rating Scale and for cognitive function, results of ECGs, tests of vital signs, and measurements on the visual analog scale for sedation. The change from the baseline of the double-blind trial to endpoint, and to other time points, was calculated. Wilcoxon’s signed-rank tests were used to test changes on the Extrapyramidal Symptom Rating Scale from baseline, and a two-tailed paired t test were used for the other measurements.
Endpoint and baseline scores for the primary and secondary effectiveness parameters, except for the CGI, were compared by using the paired t test. Change scores were calculated at endpoint and at each study visit relative to the two baselines: the baseline of the double-blind study and that of the extension study. For the CGI severity scale, frequency counts were generated.
Results
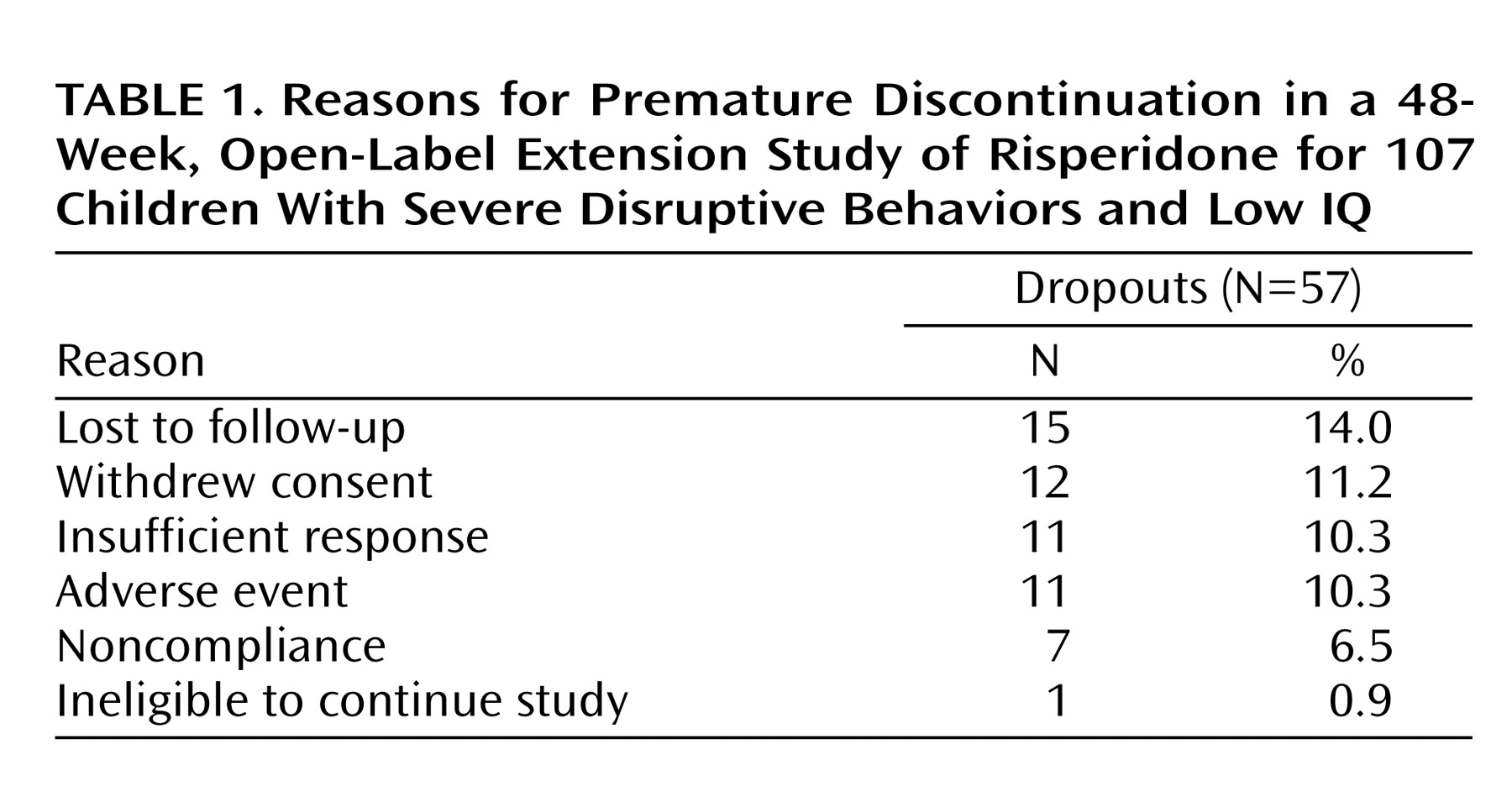
This long-term follow-up study of children with disruptive behavior disorders included 107 patients. Two patients had no assessments during the extension study and were not included in the effectiveness analysis. One patient had no effectiveness data at the baseline of the double-blind trial and therefore was excluded from the analysis of change from the double-blind baseline. Fifty-seven patients (53.3%) discontinued the extension study before its planned completion; the most common reasons were lost to follow-up and withdrawn consent (
Table 1).
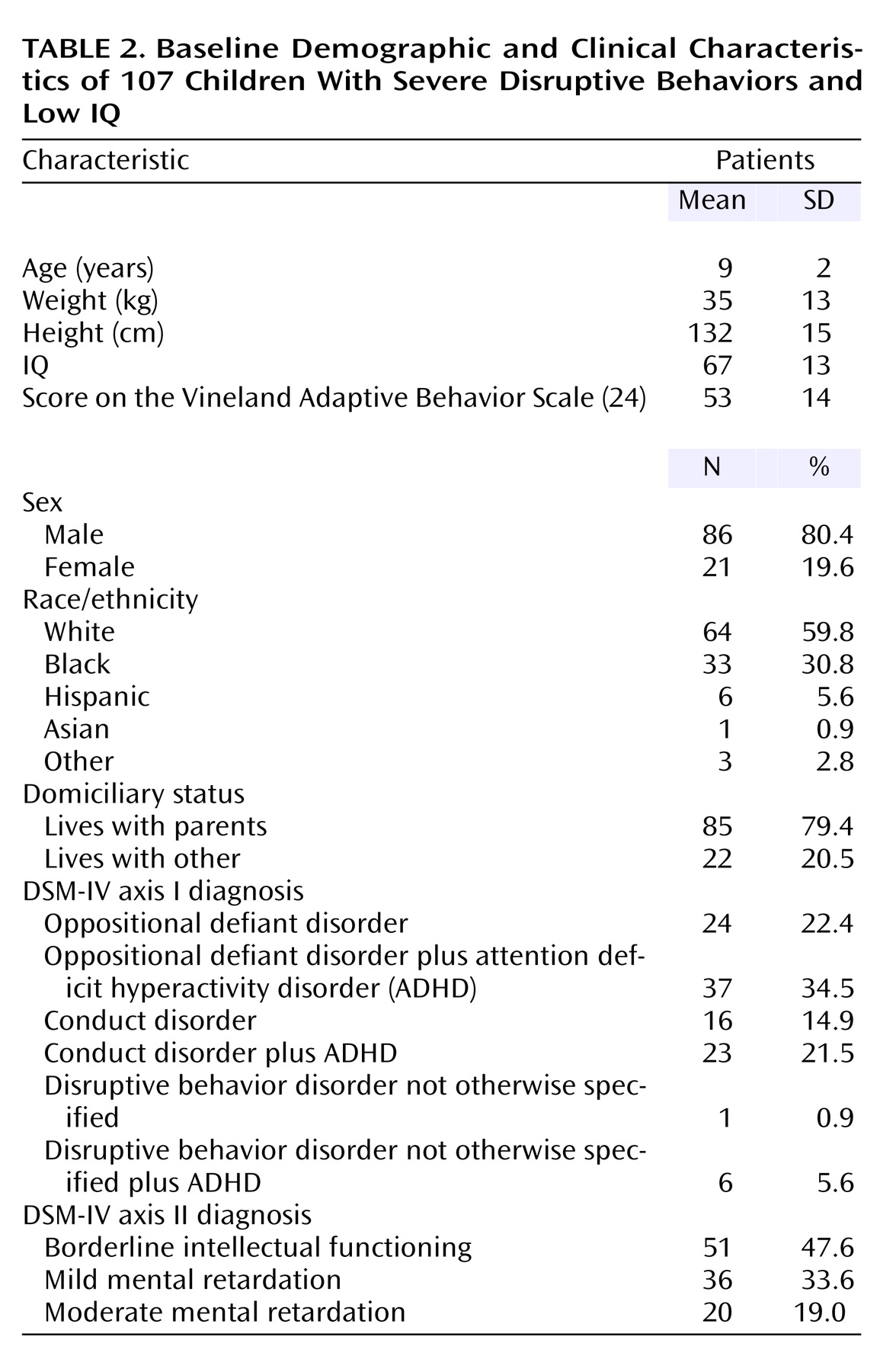
Baseline patient demographic and clinical characteristics were similar to those in the double-blind study
(19). Most patients (80.4%) were boys, and the mean age was 9 years (range=5–13) (
Table 2).
Exposure to Study Medication
The mean dose of risperidone at endpoint was 1.51 mg/day (SD=0.07) or 0.041 mg/kg/day (SD=0.001). After an initial adjustment period of approximately 4 weeks, the dose remained constant. The average treatment duration (for the acute and extension studies combined) was 34.3 weeks (SD=17.7) (range=2–388 days). Almost half of the study participants (48.5%) were also receiving psychostimulants, most commonly methylphenidate hydrochloride (30.8%).
Safety
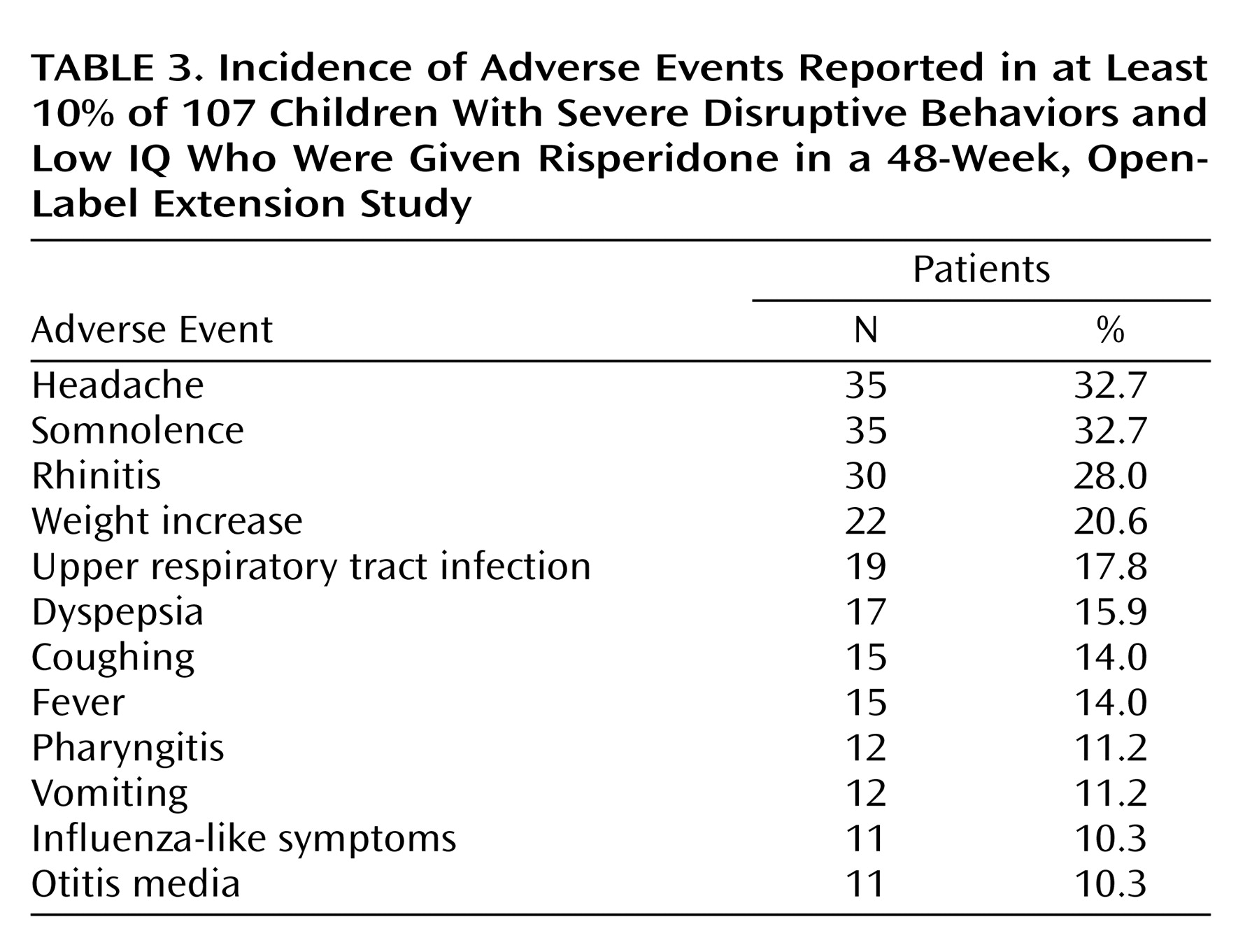
Ninety-seven patients (90.7%) had an adverse event; the most common were headache (32.7%), somnolence (32.7%), rhinitis (28.0%), and weight increase (20.6%) (
Table 3). In most cases, somnolence was mild and disappeared after a few weeks. Most adverse events were mild or moderate in severity and did not lead to discontinuation. Eleven patients (10.3%) dropped out because of adverse events. Adverse events leading to discontinuation included weight increase (N=4), depression (N=3), suicide attempt (N=2), anxiety (N=1), dystonia (N=1), headache (N=1), emotional lability (N=1), and extrapyramidal disorder (N=1). (The patients who dropped out may have reported more than one adverse effect as the reason for withdrawing.)
There were no clinically relevant mean changes in body temperature, pulse rate, respiration rate, blood pressure, ECG parameters (heart rate, P-R, QRS, QT, or QTc intervals), hematology, urinalysis, growth hormone levels, or clinical chemistry measures other than prolactin levels. Mean prolactin levels increased modestly to a maximum of 27.6 ng/ml in the boys (upper limit of normal=18 ng/ml) and 23.9 ng/ml in the girls (upper limit of normal=30 ng/ml) at week 4, with means within normal limits at endpoint (15.6 ng/ml and 16.9 ng/ml, for boys and girls, respectively). No adverse events considered to be related to hyperprolactinemia were reported during the course of the study.
During the extension trial, increased appetite was reported as an adverse event for 10 patients (9.3%) and weight gain for 22 patients (20.6%; mean increase from baseline=5.5 kg) (F=155, df=1, 104, p<0.001). One patient required medication for emergent extrapyramidal symptoms. Total scores on the Extrapyramidal Symptom Rating Scale were low at the start of the double-blind trial (mean total score=1.33) and at the start of the extension study (1.04) and remained low (0.99 at endpoint).
Most patients did not show any change from baseline on the Extrapyramidal Symptom Rating Scale at the final assessment. There was no significant change from baseline in mean scores on the dyskinesia subscale of the Extrapyramidal Symptom Rating Scale at the final assessment, and no symptoms related to tardive dyskinesia were reported as adverse events.
Scores on the 100-mm visual analog scale for sedation were low at the start of both the double-blind trial (5.0 mm) and the extension study (5.2 mm). The mean change from open-label baseline to endpoint was 8.2 mm (SD=3.3) (F=6.00, df=1, 54, p<0.02) for those who received placebo in the double-blind study and –0.02 mm (SD=2.13) (F=0.00, df=1, 47, p<1.00) for those given risperidone in the double-blind study.
Cognition
All changes on cognitive tests were in the direction of improvement over the duration of the trial. Several variables on the continuous performance task and the modified California Verbal Learning Test demonstrated statistically significant improvement over baseline at endpoint (e.g., false alarm total, F=4.2, df=1, 33, p<0.05). On the continuous performance task, errors of omission (the number of times a patient did not detect the target stimulus, indicating attentional lapses) or commission (responses to stimuli not designated as the target, indicating impulsive responses) decreased, and correct detections increased. The risperidone group and the combined placebo and risperidone groups improved significantly from open-label baseline to endpoint on the short-delay free recall portion of the modified California Verbal Learning Test (F=8.8, df=1, 141, p=0.005, and F=8.3, df=1, 84, p=0.005, respectively).
Effectiveness
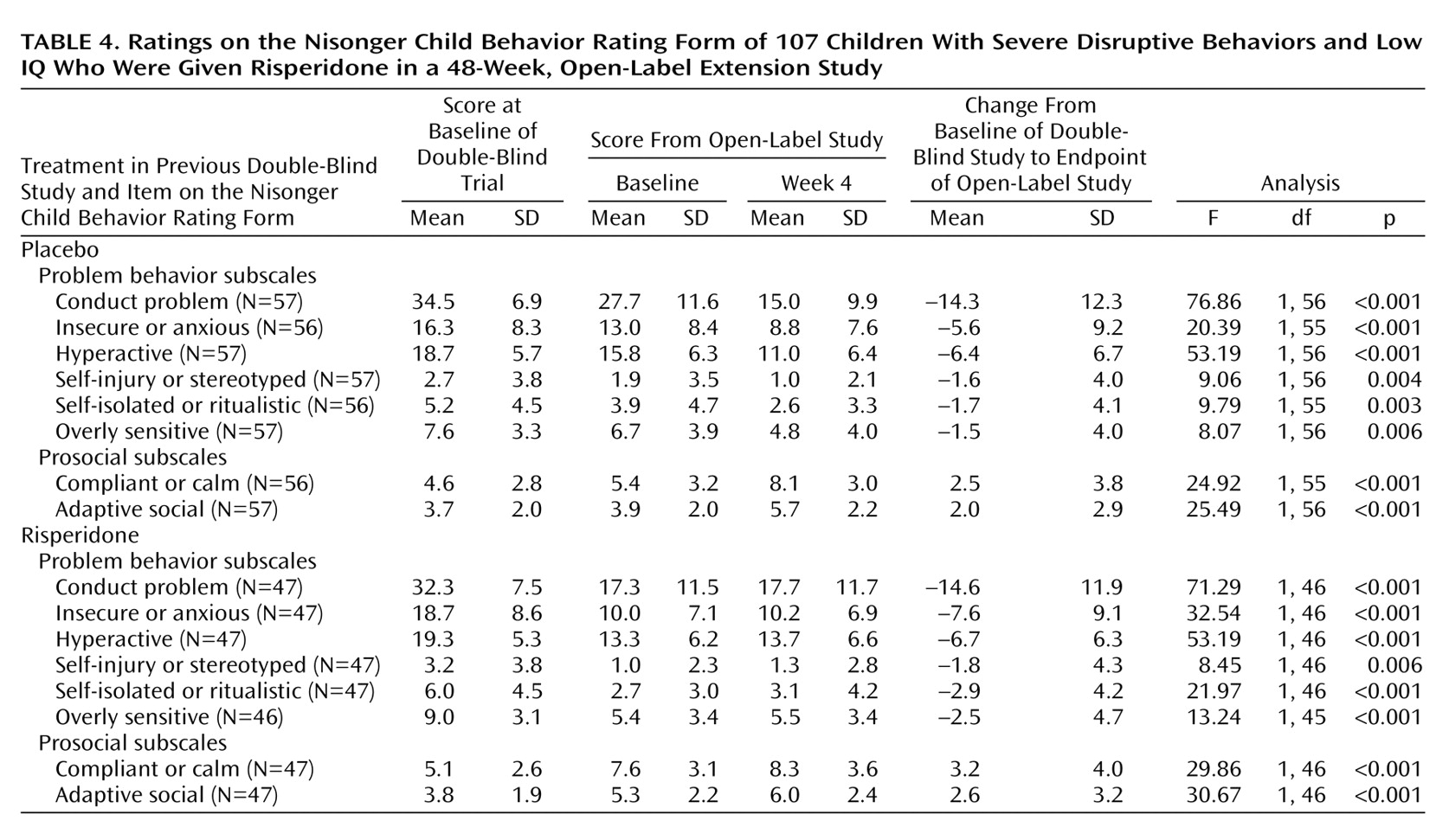
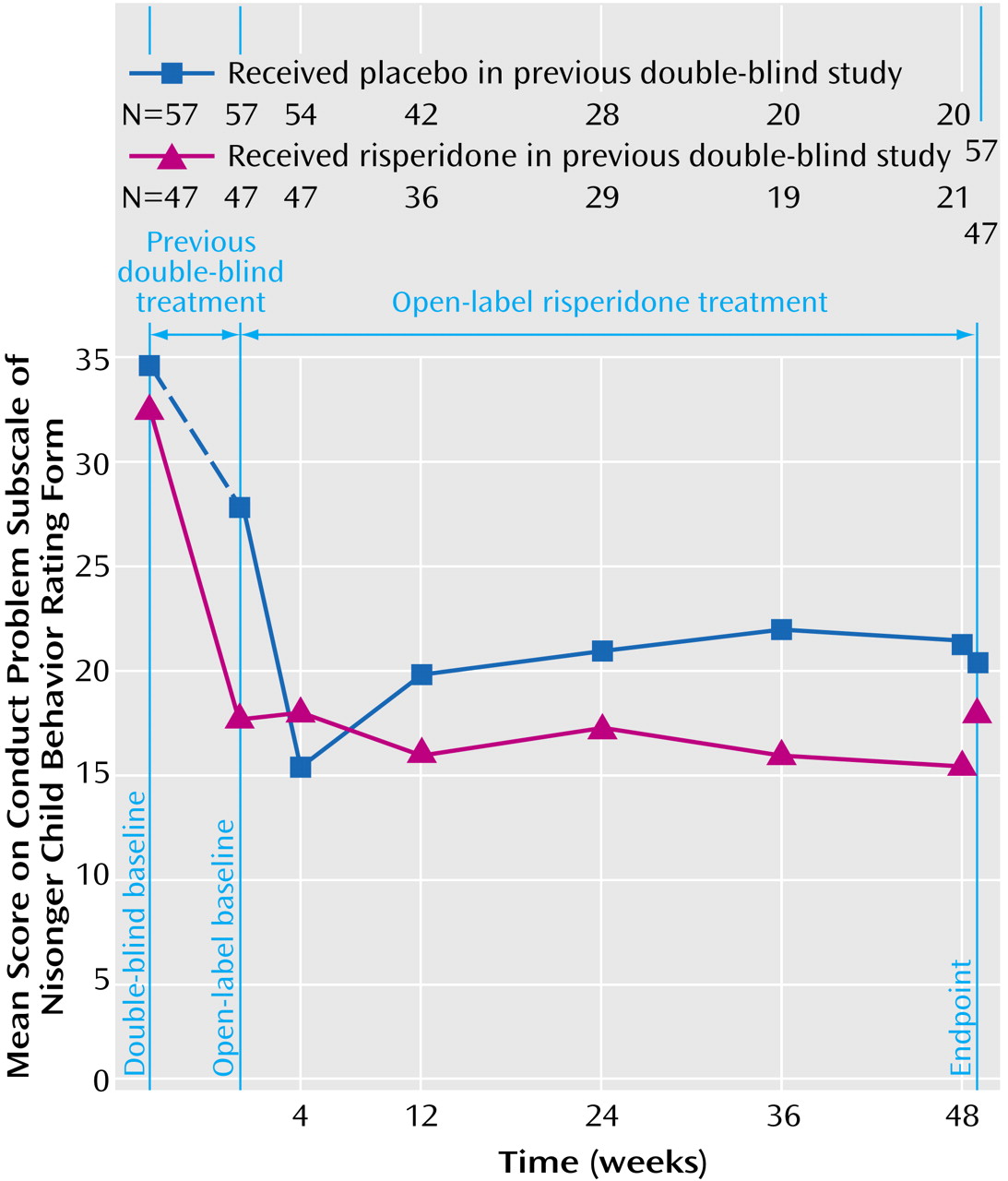
The effectiveness of risperidone demonstrated in the double-blind trial was maintained during the open-label extension among patients previously given risperidone and among those previously given placebo (
Table 4,
Figure 1). At the start of the double-blind study, baseline scores on the conduct problem subscale of the Nisonger Child Behavior Rating Form were similar in the placebo and risperidone groups (34.6 and 32.3, respectively). At the baseline of the extension study, the patients who received placebo during the double-blind trial had higher scores on the conduct problem subscale of the Nisonger Child Behavior Rating Form than did the risperidone group (27.7 and 17.3, respectively). The mean score in the double-blind study placebo group decreased during the first 4 weeks of the open-label trial (F=23.40, df=1, 56, p<0.001), such that beginning at week 4, scores in the two groups were nearly identical.
Other subscales of the Nisonger Child Behavior Rating Form also demonstrated evidence of therapeutic effectiveness. Baseline scores on these subscales were similar in both groups at the start of the double-blind trial. At the start of the extension study, subscale scores on the Nisonger Child Behavior Rating Form were higher among patients previously given placebo than among those given risperidone. In the early weeks of the open-label study, subscale scores on the Nisonger Child Behavior Rating Form in patients switched from placebo to risperidone improved and were similar in both patient groups at the end of the study (
Table 4).
Further decreases from the open-label baseline to endpoint were observed in the visual analog scale rating of the most troublesome symptom (often identified as verbal or physical aggression, hitting, fighting, or temper tantrums). Among the patients who received placebo in the double-blind study, a mean decrease of 28.5 mm (SD=31.8) from the mean open-label baseline of 66.5 mm (SD=33.1) was observed (F=26.11, df=1, 56, p<0.001). Among the patients who received risperidone during the double-blind study, a mean decrease of 8.7 mm (SD=29.9) from the mean open-label baseline of 44.3 mm (SD=32.0) was observed (F=116.37, df=1, 56, p<0.001).
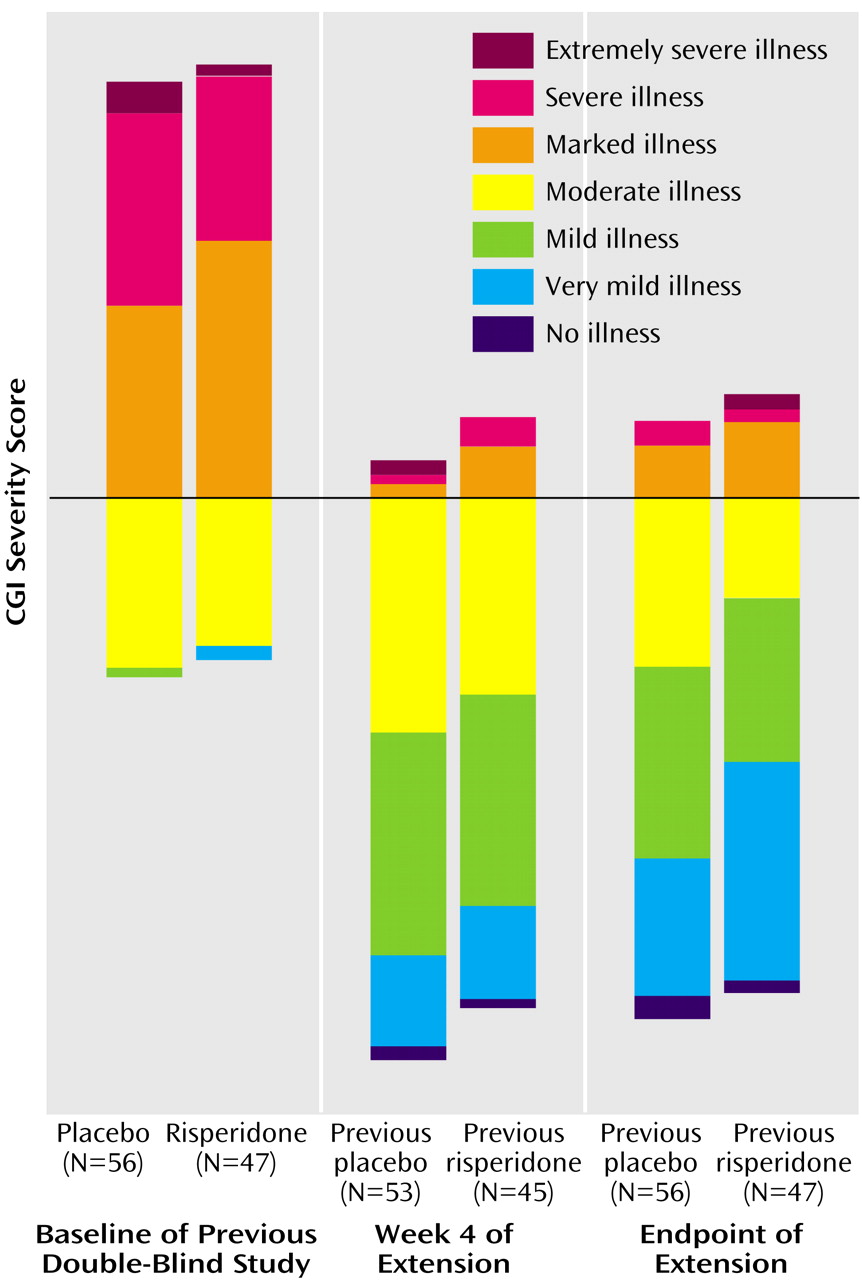
At the start of the double-blind trial, approximately 71% of the patients had CGI severity scores indicating marked, severe, or extremely severe behavioral problems. At the end of the extension trial, 62% of the patients had no symptoms or very mild or mild symptoms (
Figure 2).
Discussion
This extension study supports the findings of previous short-term studies that risperidone is safe and well tolerated and suggests that the effectiveness of risperidone in improving problem behaviors in children with developmental disabilities and disruptive behavior disorders is maintained throughout 48 weeks of treatment.
The discontinuation rate in this trial was 53.3%. This rate is not uncommon for open-label trials when the study medication is available to patients outside the trial setting. For example, the withdrawal rate in this study is similar to that reported in a long-term study of elderly patients with psychosis that was conducted when the drug was readily available outside the trial setting
(25). Moreover, the similarity in baseline characteristics of the patients who completed and who discontinued the trial make it unlikely that differences in the trial population had an influence on discontinuation rates.
Most adverse events, the most common being headache and somnolence, were mild or moderate. There were no clinically significant adverse changes in ECGs or cognitive tests. Mean prolactin levels peaked at week 4 of risperidone treatment and gradually decreased thereafter, returning to normal at endpoint. The clinical significance of transient mild temporary prolactin elevation in this study and in children in general remains unclear
(26).
Although extrapyramidal symptoms are an important risk associated with antipsychotics, they were not common in this study. At the end of the trial, mean total scores on the Extrapyramidal Symptom Rating Scale, including dyskinesia subscale scores, did not change significantly from baseline, and no new onset of tardive dyskinesia was reported. This low incidence of movement disorders is consistent with short-term, prospective, controlled studies of pediatric or adolescent patients treated with low-dose risperidone for disruptive behavior disorders in which extrapyramidal symptoms were absent or occurred no more frequently than with placebo
(6,
7). In two longer-term (6-month) studies, no dyskinesia
(13) and no extrapyramidal symptoms
(5) according to Abnormal Involuntary Movement Scale scores were observed during risperidone treatment.
In general, reports of movement disorders in children and adolescents treated with risperidone
(27–
29) have been associated with doses considerably higher (approximate mean=6 mg/day) than the dose required to achieve amelioration of target behaviors in this and other studies of this population
(6,
11,
15). A higher rate of extrapyramidal symptoms (25.9%) was, however, reported in a similar 48-week study of 77 patients with disruptive behavior disorders who were treated with low-dose risperidone
(8). Symptoms were mild or moderate in all cases, and no patients withdrew from the study because of extrapyramidal symptoms. In a 6-month double-blind, prospective trial, drooling was noted in 27% of the children treated with low-dose risperidone
(5). Although it has been suggested that drooling may be secondary to neurotransmitter blockade, it has been associated with orofacial dyskinesia in some studies. Further studies may be necessary to clarify the extent of the risk for movement disorders associated with long-term risperidone treatment in this patient population.
Mean weight at the start of the trial was 34.6 kg. which is in approximately the 90th percentile for the mean age of these patients, according to the National Center for Health Statistics. Although an approximately 4.9-kg weight gain would be expected for a child of this baseline weight and age over a 1-year period
(30), the mean weight increase in this 48-week study was 5.5 kg. Four patients withdrew from the study because of weight gain, the most common reason for withdrawal due to adverse events. Weight gain has been reported as an adverse event in other studies of risperidone
(5,
8,
17,
29,
31) and other atypical antipsychotics
(32,
33) in children with behavior disorders and other psychiatric diagnoses. Clinicians should explain to patients and parents that additional weight gain can occur during the course of risperidone treatment and should discuss the importance of regular exercise, a healthy diet, and structured mealtimes.
Although atypical antipsychotics can cause drowsiness in some patients and might, in theory, have a negative effect on cognitive function, changes on cognitive tests in this study were in the direction of improvement. It is not possible to determine whether these changes were due to practice effect, normal maturation, cognitive improvement associated with risperidone, or some combination of these effects. In addition, the lack of applicability of standard norms to this cognitively impaired population makes these findings difficult to interpret. However, the fact that there was no decrease in measures of cognitive function demonstrated in this trial is somewhat reassuring. These findings are consistent with the improvement in measures of cognitive function demonstrated in the study by Turgay et al.
(8).
On entry into the double-blind study, the children were quite symptomatic. Their mean conduct problem subscale score on the Nisonger Child Behavior Rating Form at screening was in the 99th percentile for norms for typically developing children (unpublished data from M.G. Aman) and exceeded the 97th percentile for norms for developmentally disabled children
(34). Improvements in behavioral symptoms occurred within the first weeks of treatment and were maintained throughout the duration of the 48-week trial. These findings support the durability of response demonstrated in the similar 48-week study of risperidone in children with disruptive behavior disorders and developmental disabilities
(8).
Our study has several limitations. There were many more male than female patients; however, our study population reflects the preponderance of boys among patients with disruptive behavior disorders
(35). In addition, given that an increase in height is expected in this age group, body mass index rather than body weight might have been a more meaningful measure of weight gain in this population. Although an open-label design is not optimal, the similarity of efficacy findings to those of the double-blind study in patients switched from placebo to risperidone suggests that this degree of change was robust.
The use of parental evaluation as the primary effectiveness measure in this study was also a limitation. Having input from other adults in close contact with the participants, such as their teachers, would have been helpful to confirm the changes reported by the parents.
This 48-week follow-up study suggests that risperidone is generally well tolerated at doses up to 0.06 mg/kg/day and may have long-term effectiveness in children with severe disruptive behavior disorders and subaverage intelligence. Given these findings and the chronic nature of these conditions, further study is warranted to assess the safety and efficacy of risperidone in pediatric patients treated for more than 1 year.
Acknowledgments
The investigators in the Risperidone Disruptive Behavior Study Group include R. Hagerman, M.D., Sacramento, Calif.; B. Handen, Ph.D., Pittsburgh; J. Hellings, M.D., Kansas City, Kan.; M. Lesem, M.D., Bellaire, Tex.; J. Pahl, M.D., Oklahoma City; D. Pearson, Ph.D., Houston; M. Rieser, M.D., Lexington, Ky.; N. Singh, Ph.D., Richmond, Va.; and J. Swanson, Ph.D., Irvine, Calif.