Weight gain is a commonly observed adverse effect of antipsychotic drug therapy when administered to adolescents and adults for a variety of psychotic illnesses
(1–
5). While most conventional antipsychotics produce modest increases in weight
(6), atypical antipsychotics—including clozapine, risperidone, olanzapine, and quetiapine but not ziprasidone—are associated with more marked weight gain
(7–
9). Clinical trials of the efficacy and safety of the atypical antipsychotics show weight gain in 50%–80% of subjects, ranging from a few pounds to amounts that exceed 20% of baseline weight
(3,
10,
11). Weight gain is a frequent cause of poor adherence to antipsychotic medications and a potential contributor to increased comorbidity
(12), including glucose intolerance
(13), diabetes mellitus
(14,
15), and sleep apnea
(16).
Significant weight gain is reportedly more common with clozapine and olanzapine than with other antipsychotic agents
(2,
4,
7,
17). Predictors of weight gain in olanzapine-treated subjects versus a placebo control condition include increased appetite, good clinical response, and low baseline body mass index
(9,
17,
18). These predictors are also consistent with observations from earlier studies that found an association between weight gain and good clinical response with typical antipsychotics
(19) or low baseline body mass index
(20,
21). Mechanistically, weight gain with olanzapine has been attributed to the combination of high affinities for 5-HT
2C serotonin and H
1 histamine receptors
(22). Both serotonin and histamine are involved in the regulation of food intake and can affect the level of motor activity and caloric expenditure
(23).
Despite an increased awareness of the metabolic hazards of atypical antipsychotics, many questions remain unanswered regarding the cause of weight gain induced by this group of medications and the mechanism by which it occurs. Theoretically, an atypical antipsychotic drug must trigger weight gain by inducing a positive energy balance. However, few human studies have systematically explored the effect of atypical antipsychotics on body composition and energy balance. Investigation into the impact of antipsychotic drugs on energy balance is complicated in clinical practice because most patients with psychosis have a history of multiple trials of a variety of antipsychotic medications. In addition, many patients are also being treated with multiple psychotropic medications for concomitant symptoms, and these medications (e.g., mood stabilizers and antidepressants) are frequently associated with weight gain.
The ideal population to investigate causes of weight gain from atypical antipsychotics would be a cohort of first-episode antipsychotic-naive subjects to eliminate confounding effects of other medications. Here we investigated changes in body weight, body composition, and energy expenditure in nine outpatients with first-episode psychosis before, or shortly after, they began treatment with olanzapine and approximately 12 weeks after olanzapine initiation.
Method
Subjects
Adult patients who came to The University of North Carolina Hospitals or Dorothea Dix Psychiatric Hospital experiencing their first psychotic episode were asked to participate in an investigation of olanzapine-induced weight change. Inclusion criteria were 1) age 18–65 years, 2) DSM-IV criteria met for brief psychotic disorder, schizophreniform disorder, schizophrenia, or schizoaffective disorder, and 3) no other active illness. Subjects were excluded if they had a current comorbid alcohol or drug abuse diagnosis, were pregnant or lactating, or required treatment with medications other than lorazepam, clonazepam, zolpidem, benztropine, or propranolol for any medical or other psychiatric condition. All subjects gave written informed consent for participation in the study after the procedures were fully explained and questions answered. The UNC School of Medicine Committee for the Protection of Human Subjects approved the study protocol.
Study Protocol and Outcome Measures
At screening, all subjects were assessed with the Structured Clinical Interview for DSM-IV to determine psychiatric diagnosis and eligibility for this study. Additionally, demographic information, medical history, and a physical examination were performed to rule out medical illness. The Positive and Negative Syndrome Scale was used to assess psychiatric symptom severity. Subjects who, in the judgment of the treating clinician, required medications other than lorazepam, clonazepam, zolpidem, benztropine, or propranolol to control acute agitation or anxiety prior to completion of the baseline study visit were excluded from the study.
Within 14 days of screening, subjects were admitted for a 24-hour inpatient stay at the University of North Carolina General Clinical Research Center. At this first visit, subjects underwent assessment of their body weight, waist and hip measurements, body composition, resting energy expenditure, and respiratory quotient; fasting blood studies and a urine drug screen were also performed. Olanzapine, 5 mg/day, provided free of charge was titrated by the treating physician in 2.5-mg increments, and final doses ranged between 2.5–20 mg/day on the basis of clinical response. Dose changes were made no more frequently than once in a 24-hour period. Follow-up visits to the psychiatry outpatient department of the UNC Neurosciences Hospital, the clinical research unit of Dorothea Dix Hospital, or the outpatient General Clinical Research Center occurred at weekly intervals for two visits, then biweekly for five visits. At these visits, assessments of adverse events, medication use, weight, and capillary glucose were obtained. The final study visit was conducted at the General Clinical Research Center during a 24-hour inpatient stay after the subject completed 12 weeks of olanzapine treatment. Two subjects ended the study prematurely, one at 7 weeks and the other at 10 weeks. The median time receiving treatment at last visit was 88 days (range=48–111). In addition to the aforementioned outpatient assessments, body composition, metabolic rate, and blood work were performed at this visit.
Body weight was measured with subjects wearing light clothing; a digital scale that was calibrated monthly was used. Subjects had measurements taken upon awakening during their first and last visits, and at variable times of day during follow-up outpatient visits. Height was assessed at first visit and last visit by a wall-mounted stadiometer. Waist circumference was measured at the umbilicus and hip measurement at the fullest part of the hip. Body composition (fat mass, fat-free soft tissue mass, and bone mineral content) was assessed by using dual-energy x-ray absorptiometry with a Delphi-W instrument (Hologic, Inc., Waltham, Mass.) at the first visit and last visit (approximately 12 weeks). Substrate utilization (respiratory quotient) and resting energy expenditure were determined by indirect calorimetry with a CPX/D MedGraphics Metabolic Cart (St. Paul, Minn.) at least 2 hours postprandial and after at least 30 minutes in the supine resting position. Measures of oxygen consumption and carbon dioxide production were taken at 30-second intervals over 20 minutes under standard temperature, pressure, and humidity. Resting energy expenditure was calculated using Weir’s formula
(24).
Standard laboratory methods were used to determine 1) basic chemistries; 2) fasting levels of glucose, insulin, total cholesterol, high density lipoprotein (HDL) cholesterol, and triglycerides; and 3) levels of C-peptide and prolactin. Low density lipoprotein (LDL) cholesterol was determined by the Friedewald et al. calculation
(25): LDL=total cholesterol – (HDL + [triglycerides/5]). Leptin was measured by using the Linco human ultrasensitive radioimmunoassay kit (St. Charles, Mo.). All laboratory measurements were obtained at first visit and last visit (approximately 12 weeks apart).
Data Analysis
The study’s main outcome measures were change in body weight, body composition, energy expenditure, and respiratory quotient after 12 weeks of treatment with olanzapine. For all outcomes, change was computed as the difference from first to last observation. Due to the skewed distribution of outcome variables and small sample size, nonparametric testing procedures were used. As a result, median changes are reported instead of mean changes. The Wilcoxon signed rank test was used to test whether outcome measures at last observation were significantly different from initial observation. Spearman rank correlations were used to assess association between changes in outcome variables from first to last observation. Exploratory data analyses were also performed on related variables and laboratory measures. Computations were performed using SAS
(26).
Discussion
The results of this olanzapine study are consistent with other studies that have demonstrated significant weight gain in psychotic patients treated with this class of drugs. The weight gain in this study occurred in all but two subjects. One volunteer was nonadherent to olanzapine treatment, and the results of a urine drug screen revealed the presence of cocaine, an appetite suppressant
(28). The other subject was receiving a low dose of olanzapine (2.5 mg/day); all others were receiving at least 5 mg/day. Although suggestive, this study is too small to determine if there is an association between weight gain and drug dose (or at least a threshold dose above which weight gain can occur). Other studies have attributed the weight gain to the fact that patients were returning to their premorbid weight. We did not find that subjects with self-reported weight loss before study entry, or subjects with the lowest body mass index, were any more prone to weight gain than subjects who were near their usual or peak adult weight. However, we did not confirm whether these self-reported weights were accurate. It is encouraging to know that no subject became obese in this study, but the study was of limited duration. Other studies have described a pattern of weight gain that extends beyond 6 months of olanzapine therapy
(17). We observed no clear evidence that the weight gain was tapering off after 12 weeks of treatment.
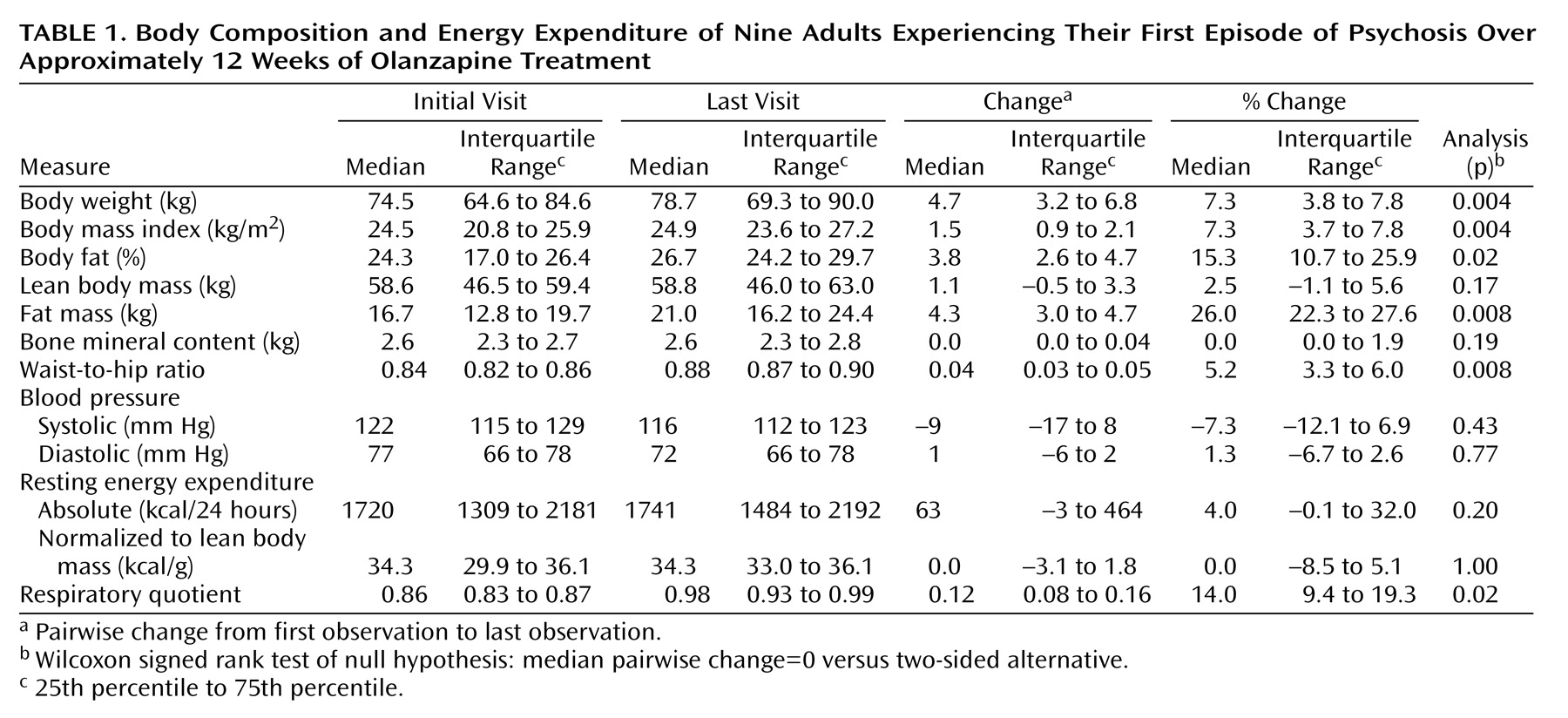
This study demonstrated by dual-energy x-ray absorptiometry that the weight gain was primarily due to an increase in body fat, which amounted to about 74% of the total weight gain. Furthermore, the increase in the waist-to-hip ratio suggests a central fat deposition, a pattern associated with adverse metabolic consequences
(29). The increase in body fat also indicates that the weight gain was not due to fluid accumulation, as is seen in treatment of diabetics with thiazolidinediones
(30). The most likely explanation for the fat gain is that subjects maintained positive energy balance and deposited this energy in the form of triglyceride in adipose tissue. From the amount of weight gain over the 12-week study period, we would have had to detect a 475 kcal/day increase in caloric intake, assuming energy expenditure did not change. In this small sample, we did not have the power to detect such a change. In addition, long-term assessment of caloric intake has multiple limitations. For example, studies using doubly labeled water to measure 24-hour expenditure have determined that obese subjects underreport caloric intake by up to 40% and overreport physical activity
(31). Similar validation studies have not been reported in psychotic patients, who often have at least mild cognitive impairment as part of their illness. Validated measurements of food intake, based on 24-hour energy expenditure, were not performed in the current study because doubly labeled water was unavailable during the study period.
It is possible that the olanzapine-induced weight gain was due to a decrease in energy expenditure. We did not see a change in absolute resting energy expenditure or resting energy expenditure normalized to lean body mass. Therefore, a decrease in resting energy expenditure is not a viable explanation for olanzapine-induced weight gain. The most variable portion of energy expenditure is physical activity. Studies in 5-HT
2C receptor knockout mice with late-onset obesity show leptin-independent hyperphagia and hyperactivity but reduced energy cost of physical activity
(25,
32). If the animal studies translate to humans, then it is possible that inhibition of this receptor subtype by olanzapine may produce similar changes in energy balances to increase body fat. In this study, efforts were made to assess physical activity with an accelerometer, but interpretation of the data was problematic, since several subjects removed the device during waking hours. However, there were no reported complaints of sedation to suggest that the weight gain was due to lack of spontaneous activity.
This study suggests that olanzapine induced a decrease in fatty acid oxidation. Decreased fatty acid oxidation has been associated with weight gain
(33). One group reported that in a study of Pima Indians, high respiratory quotient/low fatty acid oxidation was a predictor of weight gain over the long-term
(34). The decision to use fatty acids or carbohydrates as fuels is highly regulated and depends on numerous exogenous and endogenous factors, including the level of feeding (positive versus negative energy balance), the composition of food eaten (high versus low carbohydrate), the size of the glycogen stores, and the amount of adipose tissue as well as genetic factors (reviewed in reference
35). In this study, we cannot determine precisely how olanzapine induced the shift in nutrient utilization away from fatty acids toward carbohydrates, but the increase in body fat and triglyceride and insulin levels may be a reflection of this process
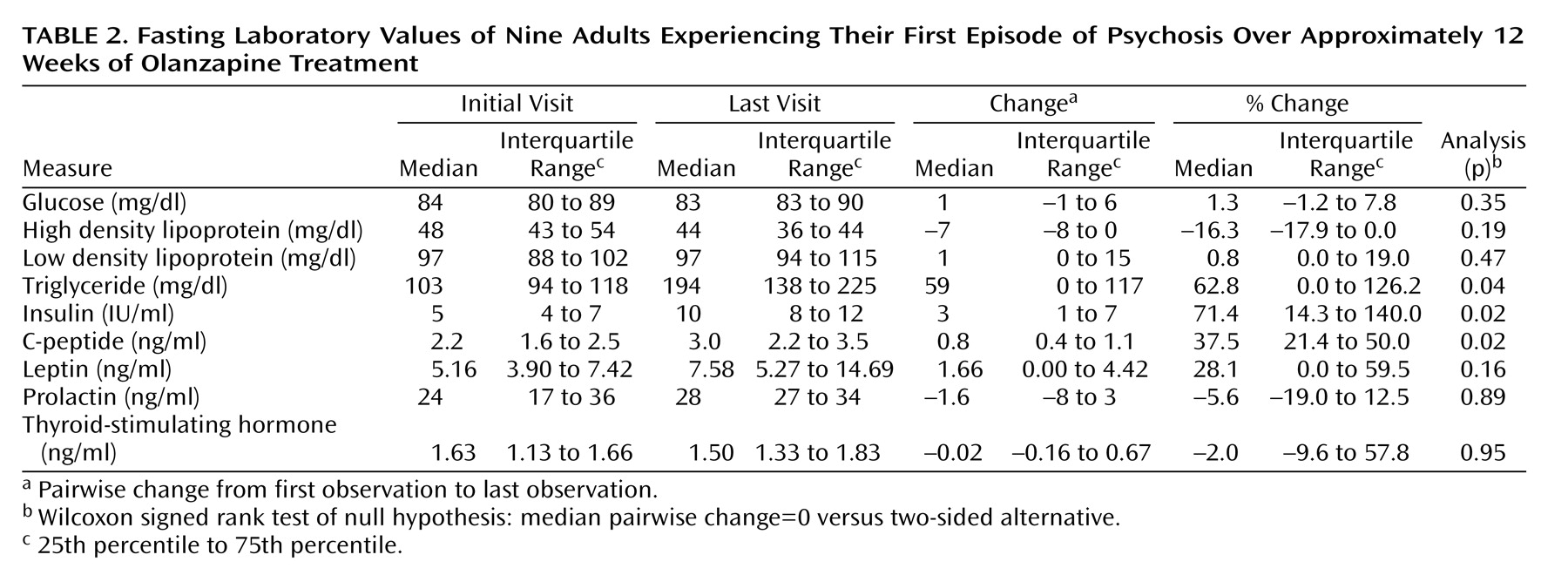
Leptin is an adipocyte-derived negative regulator of food intake
(36) that increases with adiposity
(37). Low plasma leptin concentrations have been shown to predict weight gain in some studies
(38) but not all
(39–
41). One group reported a significant mean increase in leptin levels in olanzapine-treated inpatients over a 4-week period (from 6.1 [SD=6] to 10.1 [SD=5.4])
(42). Another group found an inverse relationship between clozapine-induced increase in circulating leptin at 2 weeks and weight gain at 6 and 8 months. Here, levels of leptin were similar to those reported in the study of Kraus et al.
(42). Additionally, we detected a tendency for leptin levels to increase, but the median change with olanzapine treatment was not significant. Unlike Monteleone et al.
(43), we did not find an inverse relationship between baseline leptin levels and weight gain. This difference between our studies may be drug related, i.e., olanzapine versus clozapine. Alternatively, we compared pretreatment leptin levels to weight gain at 12 weeks, whereas they compared leptin levels after 2 weeks with weight gain at 6 months.
Atypical antipsychotics have been found to induce impaired glucose tolerance, type 2 diabetes, and diabetic ketoacidosis
(13–
15). In this study, no subject developed diabetes, but the increases in insulin and C-peptide levels were consistent with a possible decrease in insulin sensitivity. The tendency for high density lipoprotein levels to decrease and the significant increase in triglyceride levels are also consistent with the development of insulin resistance. One recent study demonstrated a decrease in insulin sensitivity in healthy volunteers treated with olanzapine or risperidone for 15–17 days
(44). However, these changes in insulin were thought to be secondary to the drug-induced weight gain. The small study group size and uncontrolled study design limit our ability to define the mechanistic pathways that led to the drug-associated weight gain and changes in glucose and lipid metabolism.
In conclusion, this study indicates that after approximately 12 weeks of olanzapine therapy, there is a significant increase in weight, increase in central adiposity, and decrease in fat oxidation. In addition, the laboratory changes are consistent with a decrease in insulin sensitivity that could lead to the development of insulin resistance syndrome. However, the size and design of the study limit the interpretation of these results. Future studies are warranted to confirm these findings, determine if these changes occur across the class of atypical antipsychotics, and determine the mechanistic basis of these clinical effects and their potential for contributing to the development of comorbid illnesses.