Major depressive disorder is a serious disorder in the pediatric age group, with 2%–8% of children and adolescents afflicted
(1,
2) . Youths with depression often have significant impairment in relationships, school, and work and are at increased risk for substance abuse, attempted and completed suicide, and depression in adulthood
(1,
3,
4) . Furthermore, it appears that early-onset major depression may be a more chronic and recurrent disorder than depression that begins in adulthood
(1,
5) . As many as 50%–75% of children with major depression have recurrent episodes
(1,
3) . Recurrence most often occurs within 6–12 months after remission
(6 –
8) . Thus, depression is a serious disorder requiring early intervention.
Treatment for major depression may be divided into three phases: acute, continuation, and maintenance treatment. Acute treatment refers to initial treatment designed to achieve response (a significant reduction in depressive symptoms) and ultimately remission (minimal or no symptoms). The goal of treatment is remission, although most randomized controlled trials include response as the primary aim. Continuation treatment follows acute treatment with the goal of preventing relapse of symptoms from the treated episode and consolidating symptom improvement for a longer duration (recovery). Continuation treatment generally lasts 4–9 months after remission. Maintenance treatment, which lasts 1–3 years, is aimed at preventing new episodes or recurrences of depression in patients who have recovered from their index episode
(9 –
11) .
Because the efficacy of antidepressants in acute treatment has only recently been established in children and adolescents
(12,
13), research on how long to continue medication treatment after response in this patient group is limited. Placebo-controlled continuation studies with adults have shown that continued treatment with an antidepressant for 6–9 months after acute treatment reduces relapse rates compared with placebo
(14 –
16) . In a small pilot study conducted as part of a large acute efficacy trial of fluoxetine in children and adolescents, continued fluoxetine reduced relapse rates compared with placebo (34% and 60%, respectively) and lengthened the time to relapse
(17) . The study was limited by several factors: it was part of a double-blind acute study; it was a small sample (N=40); randomization occurred at baseline of acute treatment, so age groups were unbalanced in the treatment groups during continuation treatment; and the length of treatment prior to randomization included both acute treatment (9 weeks) and some continuation treatment (10 weeks).
Here we present the results of a randomized, placebo-controlled discontinuation trial to evaluate the need for continuation treatment in depressed children and adolescents who responded to 12 weeks of treatment with fluoxetine.
Method
This was a single-site, double-blind, randomized discontinuation trial funded by the National Institute of Mental Health (NIMH) from August 2000 to July 2006. After a 2-week, three-visit evaluation period, participants who met all inclusion criteria and no exclusion criteria were enrolled in a 12-week open-label acute treatment period with 10–40 mg of fluoxetine. Those who responded at the end of 12 weeks of acute treatment were randomly assigned to receive fluoxetine or placebo for an additional 6 months.
The study was approved by the University of Texas Southwestern Medical Center Institutional Review Board. All participants and their parents provided written informed consent or assent after the purpose, procedures, risks, and benefits of the study and the rights of study subjects were explained and all questions were answered.
Participants
Participants were recruited from clinical referrals to a general child and adolescent psychiatry outpatient clinic as well as through advertisements. Generally, inclusion and exclusion criteria were the same as in our two previous acute double-blind, placebo-controlled trials of fluoxetine
(18,
19) . Participants were outpatients 7–18 years of age who had a primary diagnosis of major depressive disorder for at least 4 weeks, with a Children’s Depression Rating Scale—Revised (CDRS-R;
20 ) score ≥40 and a Clinical Global Impression (CGI;
21 ) severity score ≥4. Major depressive disorder had to be the primary cause for dysfunction in participants, although patients with concurrent disorders, such as anxiety, attention deficit hyperactivity disorder (ADHD), and conduct disorder, were included in the study. Participants were in good general medical health and of normal intelligence. Exclusion criteria included a lifetime history of any psychotic disorder (including psychotic depression), bipolar disorder, anorexia nervosa, or bulimia; alcohol or substance abuse within the previous 6 months; a concurrent medical condition that would interfere with the study or endanger the participant; first-degree relatives with bipolar I disorder; severe suicidal ideation requiring inpatient treatment; previous failure of or intolerance to fluoxetine; or concurrent psychotropic medications other than stimulants in a stable regimen. In females, pregnancy, lactation, and not using adequate contraception were also exclusion criteria.
Because the duration of the protocol (9 months) precluded withholding appropriate treatment for ADHD, participants were allowed to be on stimulant treatment or to begin stimulant treatment during the acute phase of the study. However, the addition of stimulant treatment was not allowed at the time of randomization or during continuation treatment.
Evaluation
Patients referred to the study were screened by telephone for possible inclusion. Appropriate subjects were scheduled for an initial diagnostic interview. After the informed consent procedure was completed, interviewers used the Schedule for Affective Disorders and Schizophrenia for School-Age Children—Present and Lifetime Version (K-SADS-PL)
(22), interviewing parents and patients separately to determine whether patients met inclusion and exclusion diagnostic criteria. The interviewer also used the Family Global Assessment Scale (D. Mrazek, unpublished, 1992) to obtain information about family functioning and the Family History Research Diagnostic Criteria
(23) to obtain a family psychiatric history. A week later, participants were evaluated by a psychiatrist or licensed psychologist. Information about course of illness and depression severity was obtained through the K-SADS-PL, the CDRS-R, and the CGI severity of illness item. Participants who met all inclusion criteria and no exclusion criteria and continued to have a CDRS-R score ≥40 were scheduled to begin acute treatment within 5–10 days.
Acute Treatment
Beginning at the baseline visit (week 0), participants received 10 mg/day of fluoxetine for 1 week, and then the dosage was increased to 20 mg/day. The dosage could be increased to 30–40 mg/day after 6 weeks of treatment if there was minimal or no response (i.e., a CGI severity score ≥3). The dosage could be reduced to 10 mg/day if intolerable side effects were present.
A child psychiatrist conducted all treatment visits and completed all rating scales. Visits were weekly for weeks 1–4 and every other week until acute treatment ended at week 12. Supportive clinical management (e.g., contact with schools and referrals for treatment for family members) was provided during each visit, although no specific psychotherapy was allowed. No concomitant psychotropic medications other than stimulants were allowed during the treatment, including the continuation phase. Participants were discontinued from the study if they did not adhere to the medication regimen; nonadherence was defined as having taken <70% of pills, based on pill count, on two consecutive visits or a total of three visits during either phase of treatment. At week 12, participants who did not respond to treatment were discontinued from the study and given recommendations for further treatment.
Continuation Treatment
Participants were eligible to enter the continuation phase if they had remitted (defined as a CGI severity of illness score of 1 or 2 and a CDRS-R score ≤28) or had an adequate clinical response (defined as a CGI severity of illness score of 1 or 2 and a decrease of 50% or more on the CDRS-R score) at week 12. Before randomization, the consent process was repeated with participants and parents for the double-blind continuation phase. Participants were then randomly assigned to receive fluoxetine or placebo. Those in the fluoxetine group received the same dose they were receiving in acute treatment. In the placebo group, fluoxetine was not tapered given its long half-life. Randomization was accomplished by a computer implementation of the minimization method in order to accommodate stratification by response category (remission versus adequate clinical response), gender, and age (participants age 12 or under and those age 13 and over). At the time the study was started, these age groups were considered an appropriate division of children and adolescents. However, in 2003, the FDA recommended including 12-year-olds as adolescents, and many “adolescent” studies included 12-year-olds
(24) . Therefore, for consistency with other trials, these suggested age groups (age 11 and under for children and age 12 and over for adolescents) were used in the analyses.
During continuation treatment, participants were evaluated by the psychiatrist every other week for weeks 12–16 and monthly for weeks 16–36, with two additional visits allowed if needed. The rating instruments were administered at each visit.
Outcome Measures
The primary outcome measures for the study were relapse and time to relapse. As noted by Rush and colleagues
(25), definitions of relapse must balance refraining from declaring a minor worsening as a full recurrence while also not requiring such a high threshold for recurrence that study subjects must endure undue pain and suffering. We defined relapse as either a one-time CDRS-R score ≥40 with worsening of depressive symptoms for at least 2 weeks, or a clinician determination that there was significant clinical deterioration suggesting that full relapse would be likely without altering treatment, even if the CDRS-R score was <40. When clinical deterioration occurred, the participant could be brought in for an interim visit reassessment or could be withdrawn from study on the basis of the second definition of relapse. We also conducted secondary analyses on the more stringent relapse definition (CDRS ≥40 only).
Secondary outcome measures included depression severity as measured by the CDRS-R; the CGI severity and improvement scales (an improvement score of 1 or 2 [very much or much improved] is considered an acceptable response to treatment); and the Children’s Global Assessment Scale
(26), which measures overall functioning, with lower scores indicating greater impairment in functioning.
Safety
Adverse events were assessed at each visit through general inquiry about problems since the last visit. Serious adverse events, as defined according to FDA criteria, include adverse events that lead to death; are life threatening; require hospitalization (initial or prolonged); lead to disability, congenital anomaly, or birth defect; require intervention to prevent permanent impairment or damage; and other important medical events that require medical or surgical intervention (e.g., failed suicide attempt).
Statistical Analyses
Acute phase baseline characteristics were compared between participants who completed the 12-week acute phase and underwent randomization and those who dropped out of the acute phase. Similarly, continuation phase baseline characteristics were compared between participants in the fluoxetine group and those in the placebo group. Unadjusted relapse rates (using both the first and second definition of relapse) were compared by chi-square test. A logistic regression model was used to compare relapse rates after adjustment for the following covariates, selected prior to conducting analyses: gender, age, race (Caucasian/non-Caucasian), duration of illness episode, number of episodes, duration of illness, age at illness onset, and continuation phase baseline scores on the CDRS-R, the CGI severity scale, the Children’s Global Assessment Scale, and the Family Global Assessment Scale. Because the rate of anxiety disorders was found to be significantly different between the fluoxetine and placebo groups in analyses of baseline characteristics, the regression was rerun to include presence of anxiety disorders in the model. Presence of anxiety disorders was not a significant predictor in the model, so it was removed and the originally selected covariates were maintained. Cox proportional hazards regression models both with and without the covariates defined above were used to compare time to relapse between groups.
Results
Acute Phase
Of 331 children and adolescents evaluated, 162 were screened out. Fluoxetine was given to 169 participants; one participant was lost to follow-up and did not return for a postbaseline visit. Thus, a total of 168 youths entered acute treatment and had at least one postbaseline visit, including 80 children (ages 7–11) and 88 adolescents (ages 12–18). The mean age for the overall sample was 11.8 years (SD=2.8); 42.3% were female, and most participants (75%) were Caucasian. Most (69%) were in their first depressive episode; the severity of depression as measured by the CGI severity scale was moderate for 30.4%, marked for 56.5%, and severe for 13.1%. The mean CDRS-R score at baseline was 57.6 (SD=7.3), which is consistent with previous studies.
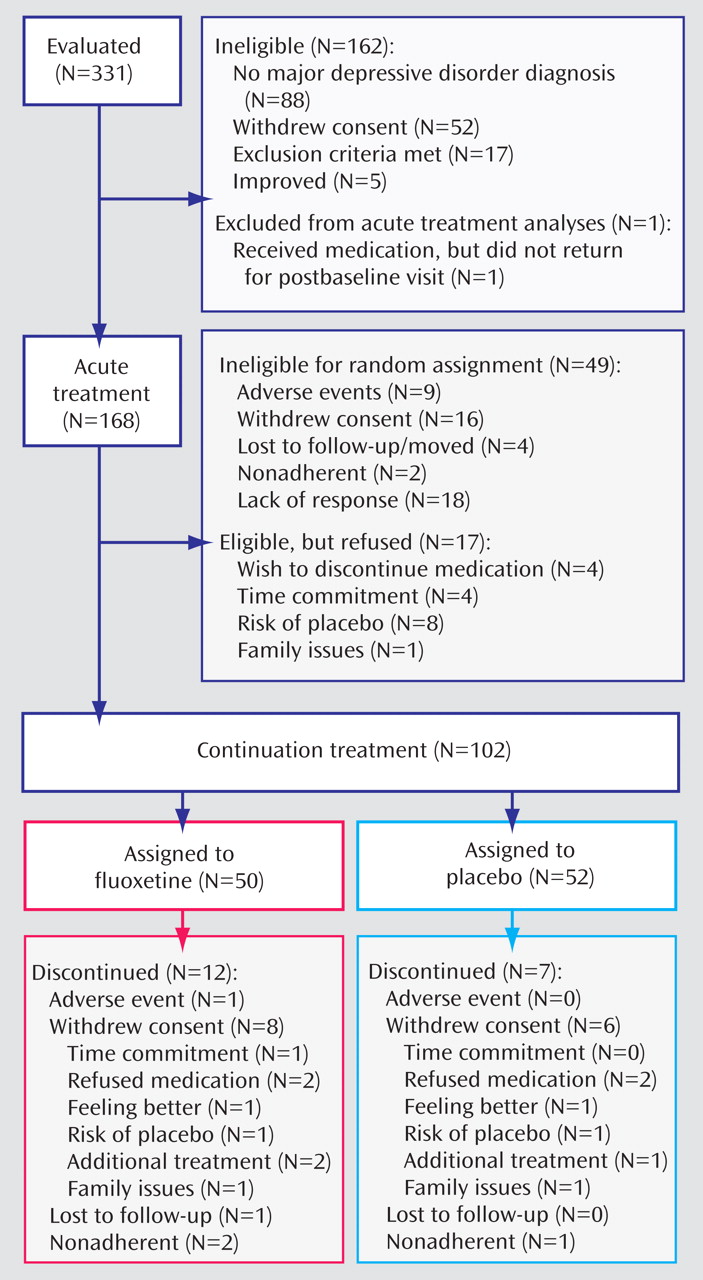
Of the 168 participants who entered acute treatment, 49 did not undergo randomization because of early withdrawal from the study or not meeting efficacy criteria; another 17 participants were eligible but did not undergo randomization. Thus, 102 participants underwent randomization for continuation treatment. The participant flow throughout the study is outlined in
Figure 1 .
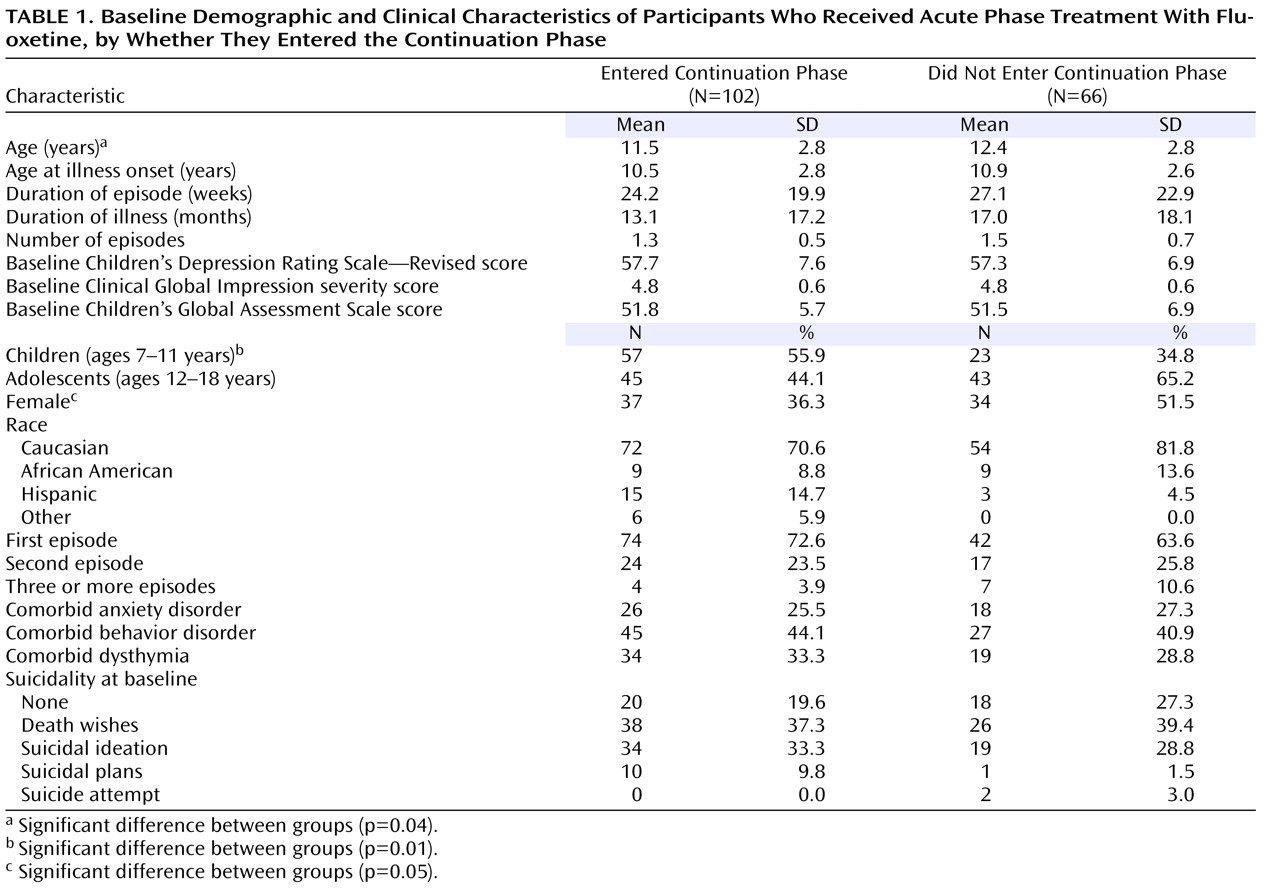
The baseline demographic and clinical characteristics of the participants who entered continuation treatment were similar to those who did not (
Table 1 ), although more participants in the younger age group entered continuation treatment compared with the older age group. In fact, of the adolescents enrolled in acute treatment, just over half (51%) entered continuation treatment, while about 71% of the children who entered acute treatment entered continuation treatment (χ
2 =7.11, df=1, p=0.01). Similarly, 52% of the females who entered acute treatment entered continuation treatment, while 67% of the males did so, although this difference did not reach statistical significance and may be confounded by age group (the adolescents were more likely to be female). Overall, illness characteristics were similar for those who entered continuation treatment compared with those who did not. Most of those who entered continuation treatment were in their first episode of depression (72.6%).
The mean dosage of fluoxetine for participants who entered continuation treatment was 26.2 mg/day (SD=9.4). The mean dosage was higher for adolescents than for children (29.8 mg/day [SD=10.1] compared with 23.3 mg/day [SD=7.9]; F=13.1, df=1, 13.1, p<0.001). Most of those who entered continuation treatment (N=70; 68.6%) had remained on 20 mg/day throughout acute treatment. The dosage was increased to 30–40 mg/day at week 6 or later in 32 participants (31.4%), most of whom were adolescents (N=22). In fact, almost half of the adolescents (48.9%) had increased to a higher dose by the end of acute treatment, while only 17.5% of the children were on a dosage >20 mg/day by the end of acute treatment. In one child, the dosage was reduced to 10 mg/day because of increased hyperactivity on 20 mg/day.
Continuation Phase
Of 102 participants who underwent randomization for the continuation phase, 50 were randomized to fluoxetine and 52 to placebo. No statistical differences were noted between the two groups with regard to age, gender, race, duration of episode, duration of illness, number of episodes, or severity of depression at study baseline. The fluoxetine group had higher rates of comorbid anxiety disorders than the placebo group (36% and 15.4%, respectively; χ 2 =5.7, df=1, p=0.02).
Participants entering the continuation phase had either remitted (CDRS-R score ≤28) or responded (CGI severity score ≤2 and a decrease of ≥50% in CDRS-R score) by the end of the acute phase. Most participants were in remission at the time of randomization (90.0% of the fluoxetine group and 86.5% of the placebo group). The mean CDRS-R score at randomization was 23.3 (SD=3.9) for the fluoxetine group and 22.4 (SD=4.4) for the placebo group. Participants in the placebo group had higher scores on the Children’s Global Assessment Scale than those in the fluoxetine group (75.7 [SD=9.3] and 71.9 [SD=8.9], respectively; F=4.38, df=1, p=0.04).
Relapse
Relapse occurred more frequently in participants in the placebo group than in the fluoxetine group (N=36 [69.2%] and N=21 [42.0%], respectively; χ 2 =7.67, df=1, p=0.009). Even using the stricter definition (CDRS-R ≥40 only), relapse was more frequent in the placebo group than in the fluoxetine group (N=25 [48.1%] and N=11 [22.0%], respectively; χ 2 =7.59, df=1, p=0.007).
In our multivariate logistic regression model examining the effect of various demographic and clinical variables on relapse rate, the treatment effect remained significant with all predictors in the model (χ 2 =5.9, df=1, p=0.0152). Given patients with median values for all covariates, the odds of relapse for the placebo group were 3.2 times those for the fluoxetine group (95% confidence interval [CI]=1.2–8.2). Similar results were obtained with the stricter definition of relapse (results not shown).
Cox proportional hazards regression showed that participants in the placebo group had a significantly greater risk of relapse than those in the fluoxetine group without adjustment for covariates (risk ratio=2.1, 95% CI=1.3–3.6; χ 2 =3.1, df=1, p=0.0044). After adjustment for the same covariates as for the logistic regression model, the risk ratio was 2.2 (95% CI=1.2–3.8) and remained significant (χ 2 =7.7, df=1, p=0.0055). Similar results were obtained with the stricter definition of relapse.
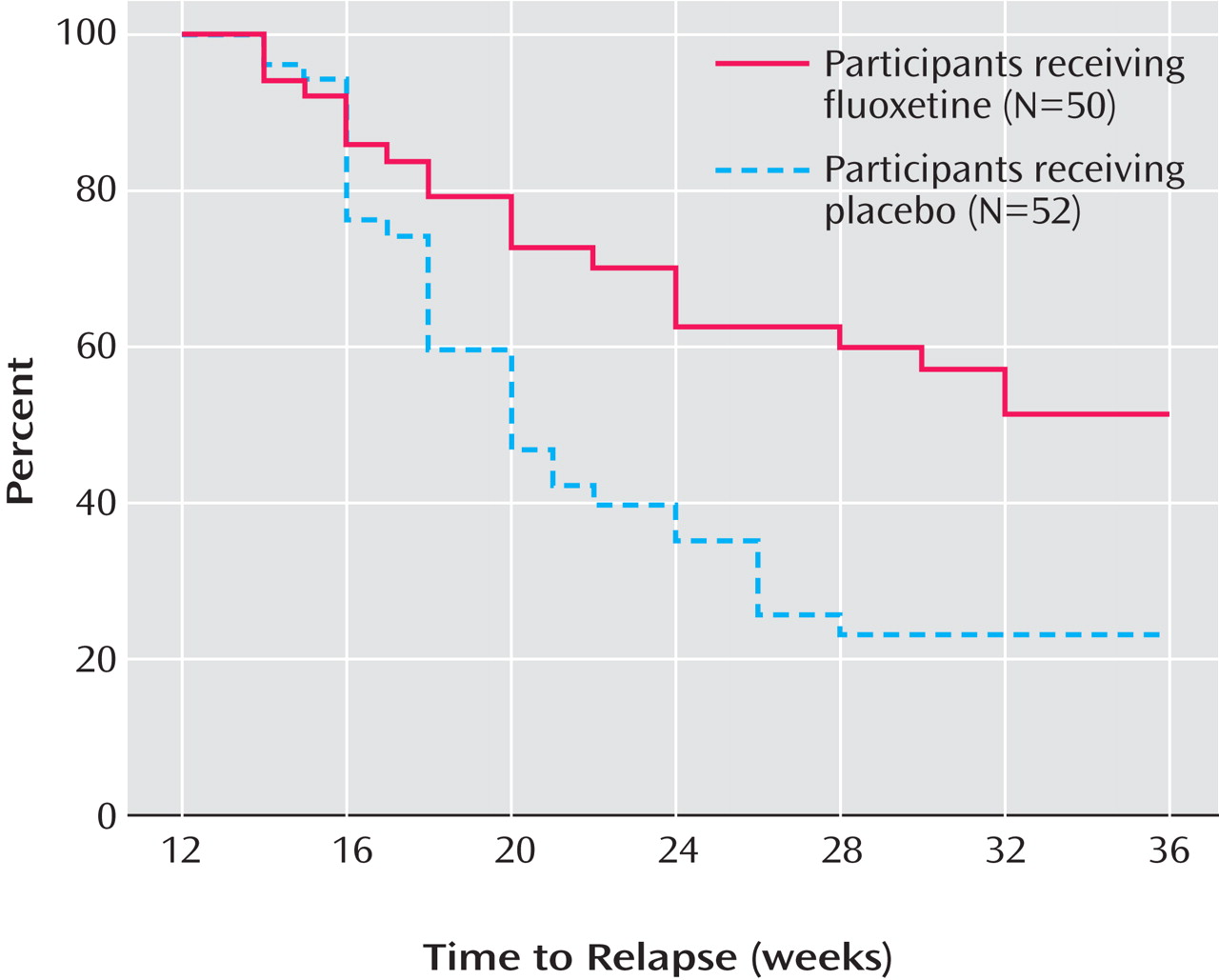
Figure 2 presents the survival curve for time to relapse, adjusted for covariates. For the placebo group, the median time to relapse was 8 weeks after discontinuation of fluoxetine. By 24 weeks after discontinuation, less than 50% of the fluoxetine group had relapsed; thus, the median time to relapse for the group could not be determined, although it is greater than 24 weeks.
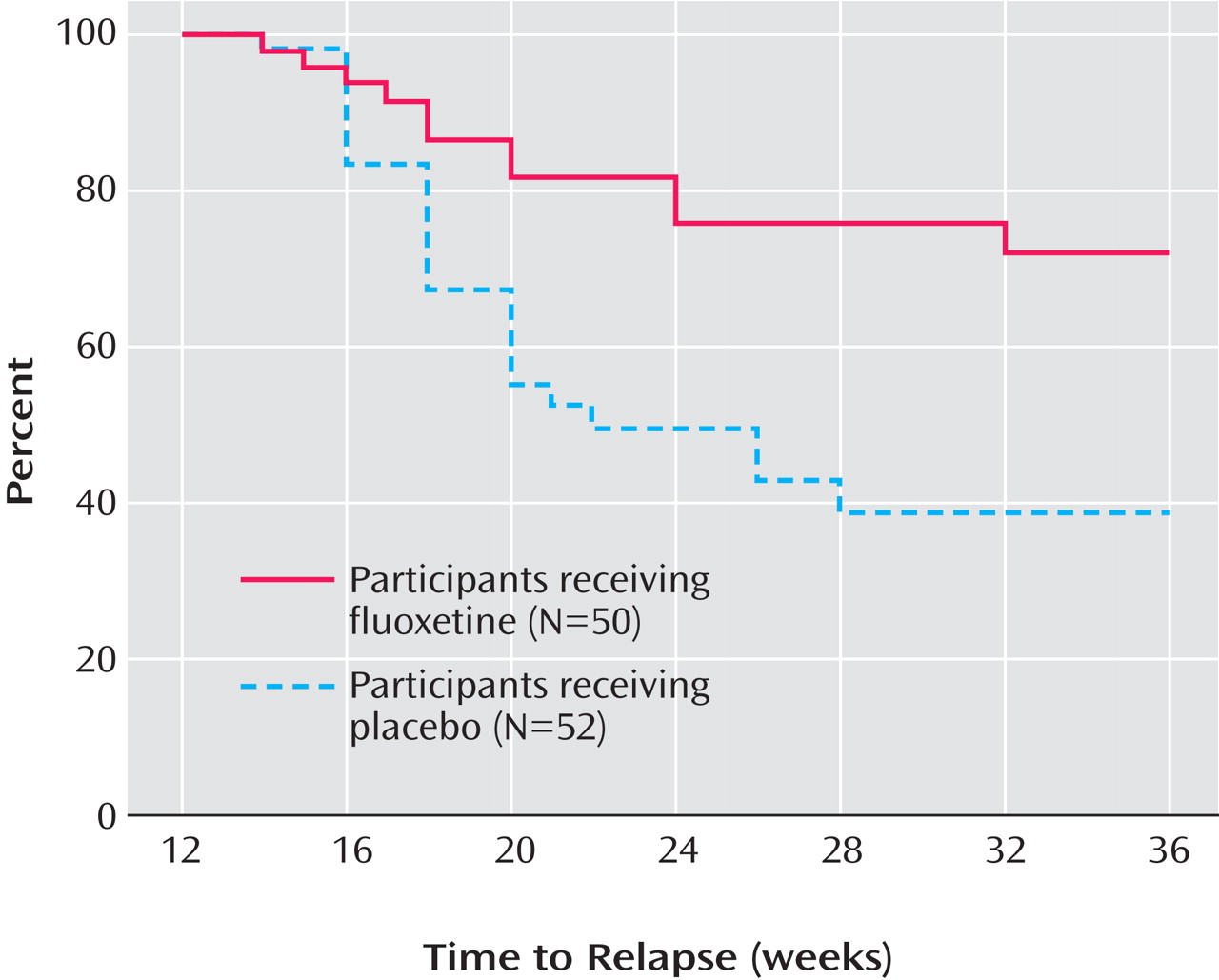
Figure 3 presents the survival curve for time to full relapse (CDRS score ≥40), adjusted for covariates. Median time to full relapse was 14 weeks for the placebo group and could not be determined for the fluoxetine group except that it is greater than 24 weeks.
Within 6 weeks of randomization, the estimated probability of relapse was 38.7% for the placebo group, compared with 19.1% for the fluoxetine group. By 12 weeks, the estimated probability of relapse was 65.7% for the placebo group, compared with 35.7% for the fluoxetine group. Only a few additional participants relapsed between 12 weeks and 24 weeks of continuation treatment in either group.
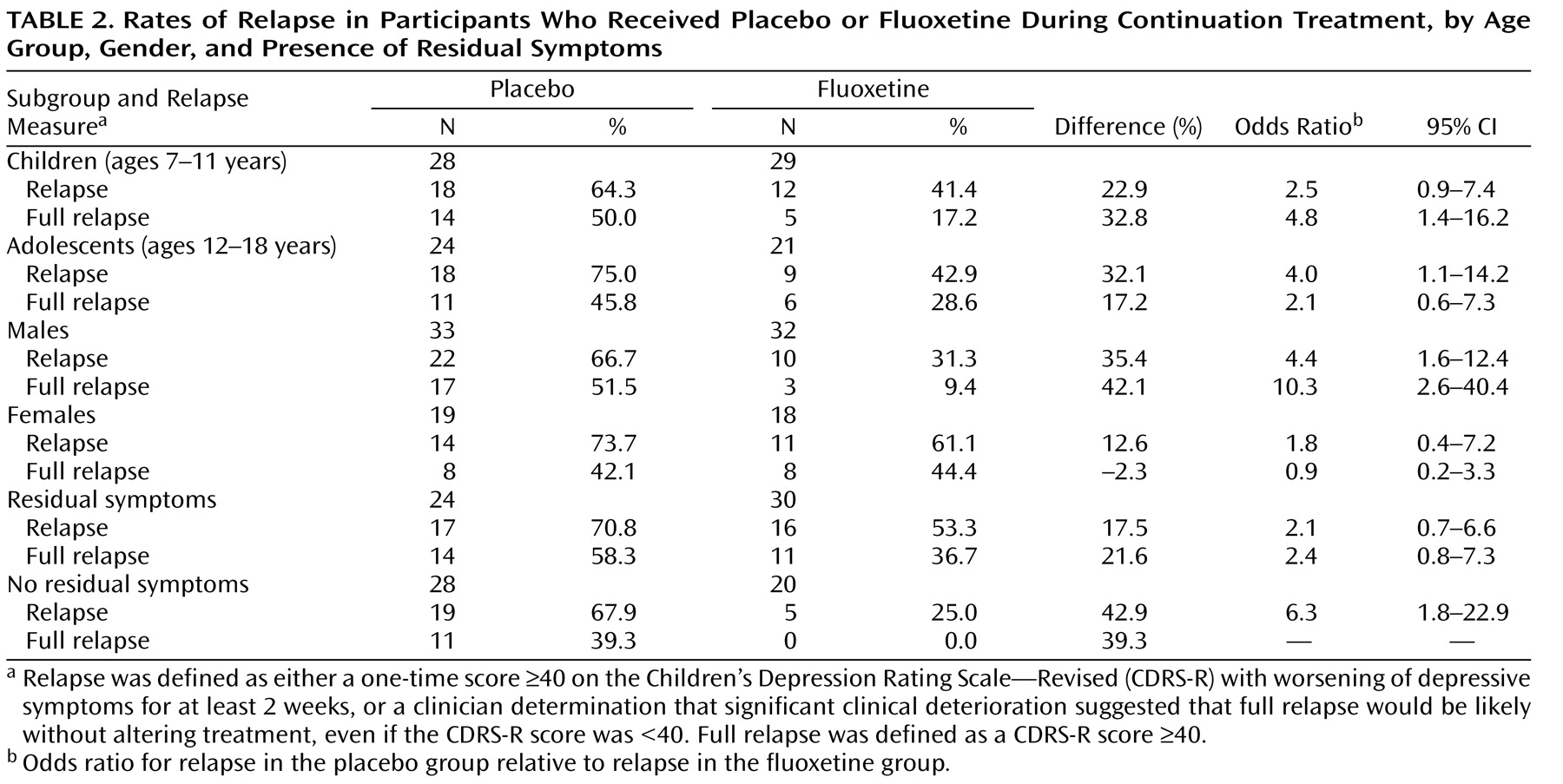
Relapse Rates by Age, Gender, and Presence of Residual Symptoms
Exploratory analyses were conducted to evaluate the effects of age, gender, and presence of residual symptoms on relapse rates (
Table 2 ). Overall, the difference between the fluoxetine and placebo groups was greatest in males when the full relapse definition was used and least in females when either definition of relapse was used. Females overall constituted a smaller group and tended to be in the older age group, which may confound results.
At the end of acute treatment, 54 (52.9%) participants reported at least one residual depressive symptom. During continuation treatment, full relapse was significantly lower in participants who had no residual symptoms at the end of acute treatment (22.9%; 11/48) than those with continued symptoms (46.3%; 25/54), regardless of treatment assignment (χ
2 =6.8, df=1, p=0.014). The greatest difference between the fluoxetine and placebo groups was observed in participants who had no residual symptoms after acute treatment (67% and 25%, respectively). Participants with no residual symptoms who were switched to placebo for continuation treatment were six times as likely as those remaining on fluoxetine to relapse (
Table 2 ).
Safety
Adverse events were similar between the two groups, and there were no discontinuations due to physical adverse events during continuation treatment. Three serious adverse events occurred during the continuation phase: two participants in the placebo group were hospitalized for preexisting medical conditions, and one participant in the fluoxetine group was withdrawn after a suicide attempt (week 16); the patient had a history of self-injurious behavior and suicidal plans without intent prior to acute treatment with fluoxetine.
Discussion
This is the first randomized, placebo-controlled study of the efficacy of continued antidepressant (fluoxetine) treatment in pediatric patients with major depressive disorder who have had an adequate response with 12 weeks of acute treatment. Fluoxetine was superior to placebo in preventing relapse and in increasing time to relapse. Relapse rates were high and occurred equally in children and adolescents. In this sample, overall relapse rates were similar in males and females, but the impact of continued treatment with fluoxetine was greater in males. Similarly, participants who had residual symptoms at the end of 12 weeks of acute treatment were more likely to relapse during the subsequent 6 months of continuation treatment on both fluoxetine and placebo. Fluoxetine was most effective for preventing relapse (compared with placebo) in those who had no residual symptoms at the end of acute treatment.
Overall, our continuation treatment sample was fairly young (mean age=11.5 years [SD=2.8]), attributable in part to the recruitment of participants in a children’s hospital. In addition, there was differential attrition for children and adolescents prior to randomization to continuation treatment; adolescents were less likely than children to remain in the study until this point. For 70% of participants, this was their first episode of major depression, and comorbid disorders were common. While participants were outpatients and therefore could not be in need of hospitalization for suicidal behavior, 43% had suicidal ideation during this episode.
Several methodological issues were raised prior to initiating the study, including concerns about the safety of discontinuation. Safeguards were recommended by NIMH, among them the exclusion of participants with a history of severe suicide attempts and the repetition of the consent protocol prior to randomization to continuation treatment. Some participants did refuse randomization (N=17 of 119 eligible, or 14.3%), about half (N=8) because they were concerned about relapse or being randomized to placebo. One concern in the planning stage was that if participants knew they were being randomly assigned to treatment, they might relapse shortly thereafter in anticipation of possibly getting placebo. However, the pattern of relapse was consistent with the half-life of the medication, so it appears this concern was unfounded.
There were substantial concerns about how to define relapse to avoid allowing participants to become too ill before declaring a relapse as well as counting minor worsening (which might improve spontaneously) as a sign of relapse. The majority of difference between the fluoxetine and placebo groups in the study was driven by the stricter definition of relapse, which required a CDRS-R score ≥40. Future studies might consider using a fairly conservative definition of relapse, although this would have to be balanced with what parents and children will tolerate.
The study is significant for several reasons. It demonstrates that continuation treatment is required beyond remission of symptoms to prevent relapse, which suggests that the adult guidelines recommending 6–9 months of overall treatment for major depression would apply equally to children and adolescents. It also reinforces the fact that early-onset depression is associated with high rates of relapse, even though the majority of participants in this sample were in their first episode of major depression.
In addition, the results support the efficacy of fluoxetine over placebo, albeit through a design not typically used to establish efficacy. The drug-placebo difference in this study was 27%, which was similar to three prior acute efficacy trials that have been published
(18,
19,
27), in which the differences ranged from 15% to 26%. As noted by Dr. Robert Temple at a recent FDA workshop on medication effectiveness (Jan. 9, 2007), it is possible that alternative designs, such as the discontinuation design used in this study, are potentially useful in establishing efficacy
(28) .
Future research would benefit by examining different treatment strategies to improve remission rates and prevent relapse. As has been observed in adults, children and adolescents with major depression who had residual symptoms after 3 months of medication and clinical management were more likely than those without residual symptoms to relapse, regardless of whether they continued medication or switched to placebo. Participants with no residual symptoms rarely relapsed on continued medication treatment. Continuation treatment with fluoxetine alone after 12 weeks is unlikely to improve remission rates
(29) . However, fluoxetine combined with cognitive-behavioral therapy (CBT) has demonstrated improved remission rates overall
(30) . As demonstrated in adults, it is possible that augmenting antidepressants with CBT after response in acute treatment could improve long-term outcome for children and adolescents, particularly for those who continue to have residual symptoms after an adequate trial of medication
(31 –
36) . Another strategy to improve remission might include medication augmentation
(37,
38) .
Treatment guidelines recommend maintenance treatment for 1–3 years for patients who have had multiple or severe depressive episodes
(3,
39,
40) . No maintenance trials have been conducted in pediatric depression; however, such trials are needed to examine the utility of long-term medication as well as to examine which patients are most likely to need extended treatment beyond continuation care.