Our group recently completed genome-wide association and candidate gene studies of nicotine dependence based on the contrast between nicotine-dependent smokers and smokers who used at least 100 cigarettes in their lifetime but never developed any symptoms of dependence
(5,
6) . These genetic studies focused on the transition from smoking to the development of nicotine dependence. Intriguing findings for further follow-up included the identification of a strong association of nicotine dependence with genetic polymorphisms in the nicotinic receptor gene cluster, α5-α3-β4, on chromosome 15, which included a variant that results in an amino acid change (aspartic acid [D] to asparagine [N]) in the α5 neuronal nicotinic acetylcholine receptor subunit (
CHRNA5 ). There was also evidence of a second distinct finding in this cluster marked by rs578776 (pairwise r
2 <0.2 with rs16969968) in the α5-α3-β4 gene cluster.
The purpose of this study was to further define the genetic contribution of these findings to smoking in families affected with alcoholism. This involved testing whether the original observations could be replicated and fine-mapped in an independent data set, examining the frequency of the variant resulting in the amino acid change in diverse populations and performing a functional study to determine whether the amino acid substitution changed receptor function.
Results
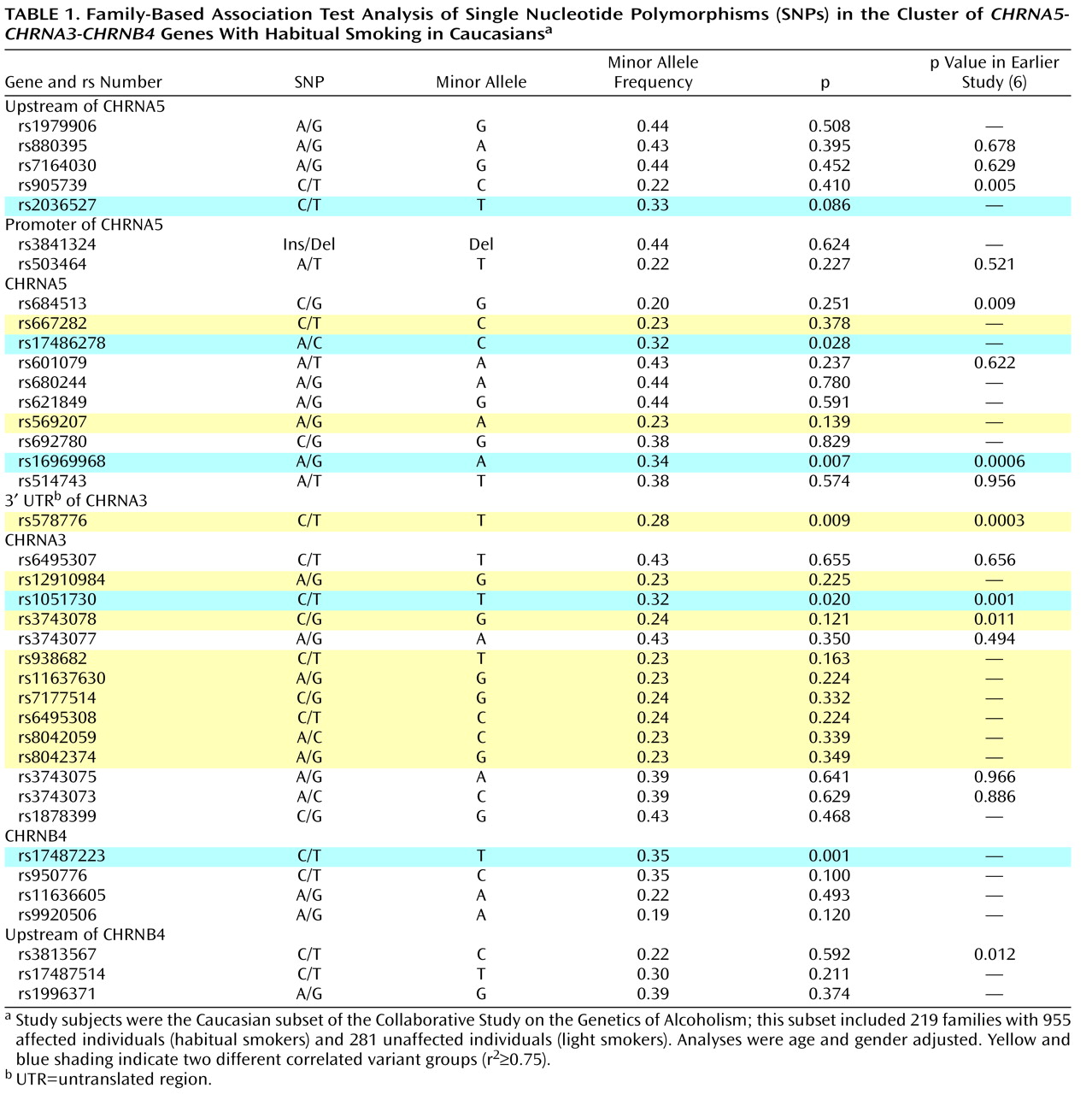
A careful examination of the linkage disequilibrium pattern across the gene cluster revealed evidence of two distinct findings of genetic association with habitual smoking versus light smoking in the
CHRNA5-CHRNA3-CHRNB4 gene cluster. The nonsynonymous coding SNP of the
CHRNA5 gene, rs16969968 (p=0.007), was associated with habitual smoking. Other SNPs that were highly correlated with rs16969968 (rs2036527, rs17486278, rs1051730, rs17487223, r
2 >0.79; see Figure S2 in the online data supplement) were also associated, with p values ranging from 0.020 to 0.086 (
Table 1 ). This SNP cluster, which spans the three genes
CHRNA5-CHRNA3-CHRNB4, most likely represents one group of correlated associated genetic variants.
A second finding of association in this gene cluster, at rs578776, was statistically independent, with a low correlation with rs16969968 (r
2 <0.15) (see
Table 1 and Figure S2). Between rs16969968 (highlighted in blue in
Table 1 ) and rs578776 (highlighted in yellow in
Table 1 ), the absolute value of D′ is high (D′=1) but in repulsion phase. The minor allele at locus rs16969968 (allele A) is on the same chromosome as the common allele at locus rs578776 (allele C), which results in the very low r
2 . Several SNPs that were moderately correlated with rs578776 (r
2 values ranging from 0.60 to 0.76) and that were significant in the previous study
(6) were not associated in this family-based analysis.
The finding of the amino acid change associated with nicotine dependence at rs16969968 was further examined in biological studies. Using sequence data from public databases, the protein sequences for
CHRNA5 homologues were aligned to determine conservation in the region surrounding codon 398 in divergent species. The aspartic acid residue at amino acid position 398 was completely conserved from human to chicken, suggesting that it has functional importance (see
Figure 1 ).
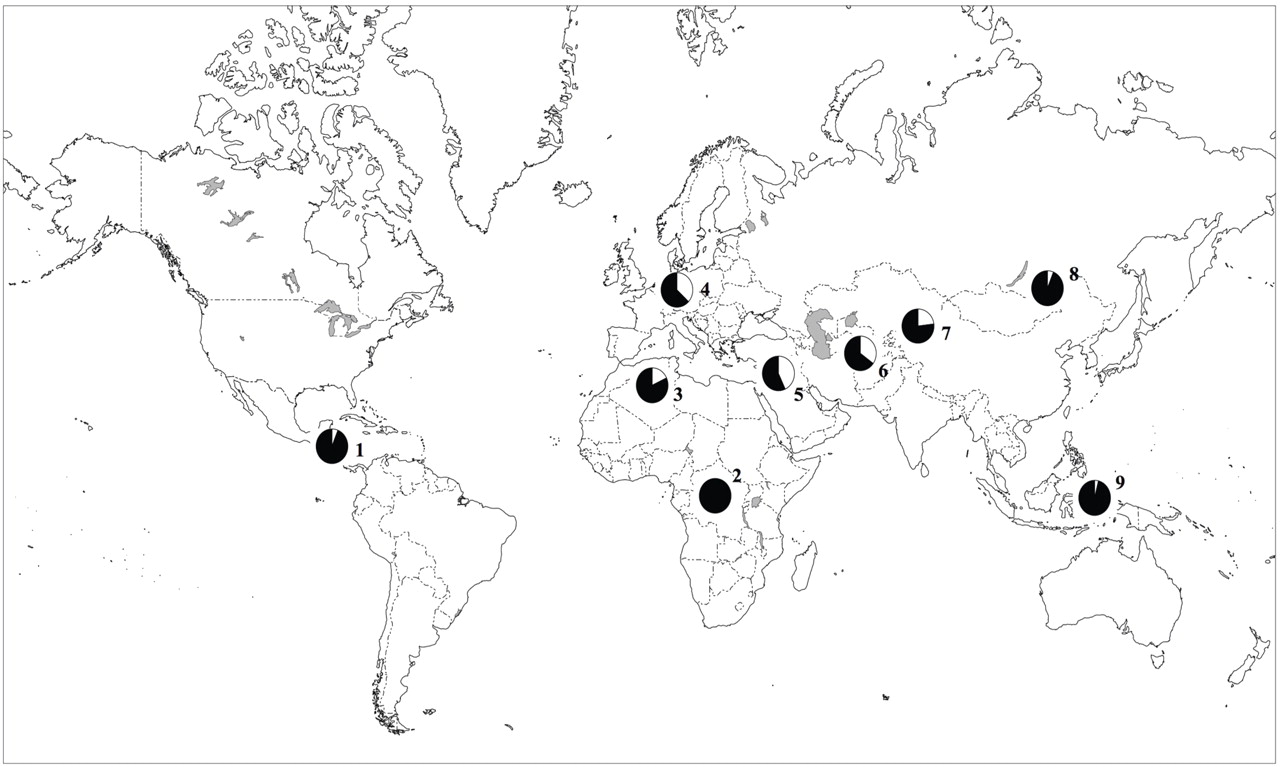
To assess the frequency of the minor allele (A) at rs16969968 across multiple populations, this SNP was successfully typed in the Human Genome Diversity Cell Line Panel, which included 995 individuals representing 39 different populations
(23) . In populations of European and Middle Eastern origin, the frequency of the A allele was 37%–43%. The A nucleotide was not detected or was uncommon in African, East Asian, and Native American populations (
Figure 2 ; see also Table S2 in the online data supplement).
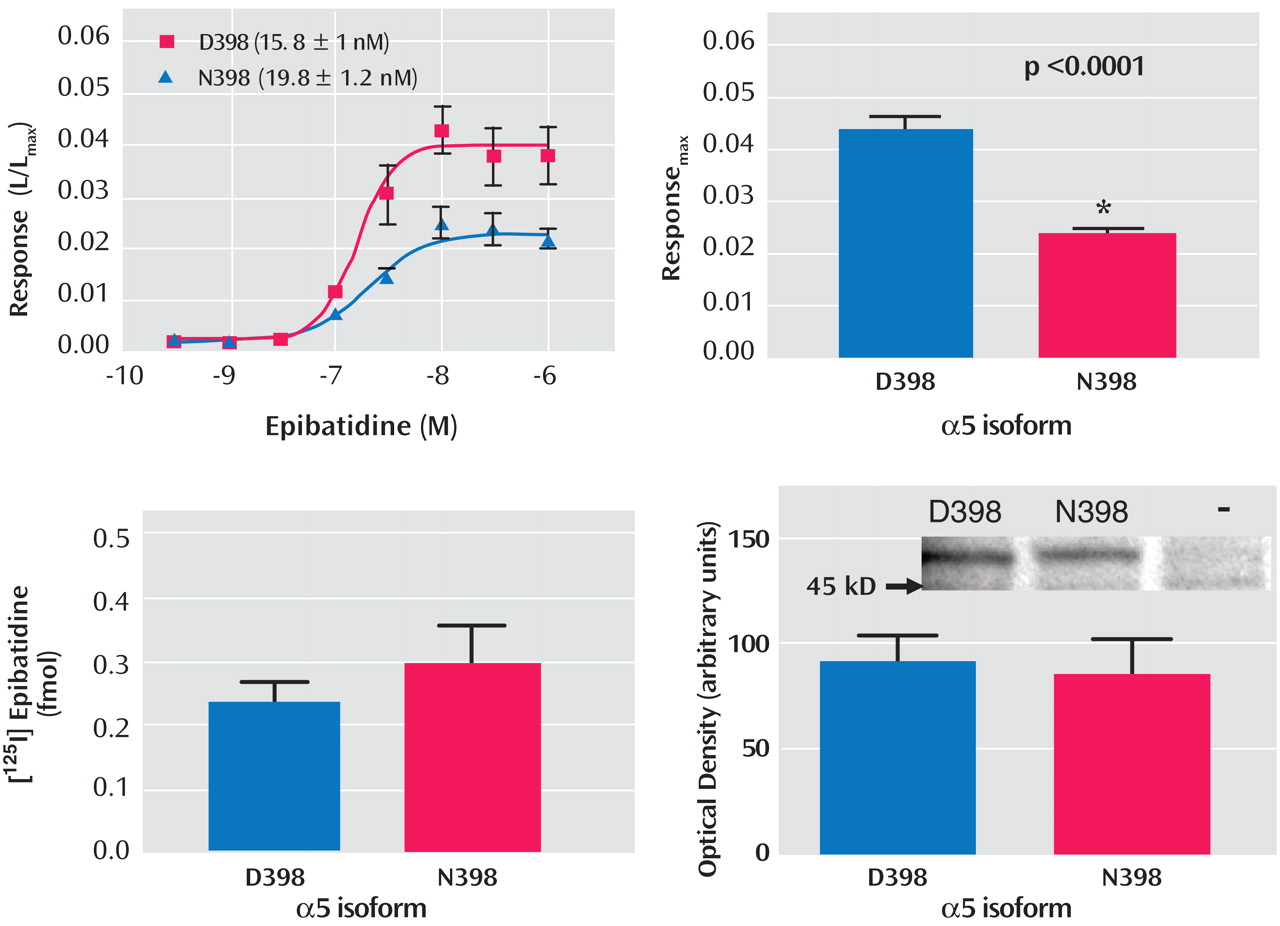
To establish whether the D398N polymorphism altered receptor function, nicotinic agonist-evoked changes in intracellular calcium were measured from HEK293T cells that heterologously expressed either α4β2α5D398 or α4β2α5N398 nAChRs. Receptor expression and α5 protein levels also were determined for each α5 variant. Two-way ANOVA indicated that the concentration response curves for the nicotinic agonist epibatidine were significantly different between the α4β2α5N398 and α4β2α5D398 nAChR variants (p<0.0001) (
Figure 3 ). This difference in concentration-response curves and maximal response to agonist was not due to a shift in sensitivity to activation by epibatidine between the nAChR variants, as their EC
50 values did not differ significantly (α4β2α5D398 EC
50 =14.7 nM [SEM=2.4]; α4β2α5N398 EC
50 =22.0 nM [SEM=3.5]; p=0.09). However, the maximal response to agonist was found to be more than two times higher for the α4β2α5D398 nAChR variant relative to the α4β2α5N398 nAChR variant (0.044 [SEM=0.002] and 0.023 [SEM=0.002], respectively; p<0.0001). In contrast, the two variant nAChRs did not differ in expression, nor were the two isoforms of the α5 subunit differentially expressed (see
Figure 3 ). The sum of these data indicates that the variant forms of the α5 subunit alter receptor function without affecting receptor expression.
Discussion
This study validates the importance of genetic variants within the α5-α3-β4 nicotinic acetylcholine receptor gene cluster that contribute to the risk of a light smoker transitioning to heavy smoking and sheds light on a potential biological mechanism. There are two distinct genetic associations—one marked by rs16969968, which results in an amino acid change in the α5 nicotinic cholinergic receptor
(CHRNA5), and a second marked by an SNP rs578776 in the 3′ untranslated region of the α3 nicotinic cholinergic receptor
(CHRNA3) . Because the r
2 between these two variants is low (0.15), the statistical significance at both cannot be explained solely by the linkage disequilibrium between them. Therefore, these data imply that two distinct loci in this region alter the risk for nicotine dependence and habitual smoking. These findings confirm our previous results in a case-control series studying nicotine dependence
(6) and the recent work by Berrettini et al. examining heavy smoking
(37) .
In these three genetic studies demonstrating the associations, there are important differences in the definition of phenotypes, recruitment procedures, and analytic methods. The COGA sample was recruited as a high-risk population for alcoholism, and a broader smoking phenotype was used: habitual smoking, defined as smoking 20 cigarettes a day for 6 months or more, and light smoking, defined as smoking 10 cigarettes or fewer per day. A family-based analytic design for association was used, which is less likely to be biased by population stratification. The previous genetic study sample our group studied was recruited from the community, the phenotypic status was defined by the Fagerström Test for Nicotine Dependence, and a case-control design of unrelated individuals was used
(5,
6,
20) . Similar SNPs were analyzed in these two studies. The recent publication by Berrettini et al.
(37) was based on genetic studies of heart disease and other common illnesses in a population-based sample of 15,000 people. Smoking status was collected, and a quantitative phenotype defined by number of cigarettes smoked per day was studied in a secondary genetic analysis. Although different genetic variants were tested in the Berrettini et al. sample, the associated SNPs are highly correlated (in linkage disequilibrium) with the variants in our studies. The consistency of results across three different studies shows that these genetic findings are robust across populations, phenotypic classification systems, and analytic methods.
Three other research groups recently published strong evidence of genetic association of the α5-α3-β4 nicotinic acetylcholine receptor gene cluster on chromosome 15 with lung cancer
(38 –
40) . The findings highlighted in these studies are highly correlated with the genetic variant that results in the amino acid change in the a5 nicotinic acetylcholine receptor gene. However, the three groups differed in their interpretation of whether this genetic association with lung cancer acts through the indirect effect of smoking or whether this variant also directly increases the vulnerability to lung cancer.
When a genetic association is found, it represents association with not only the tested variants but all genetic variants (tested and untested) that are highly correlated. Identifying the variant that causes functional changes requires biological investigations. We have focused on the amino acid change in the a5 nicotinic cholinergic receptor for further studies.
The aspartic acid at position 398 in
CHRNA5 in humans occurs at a residue that is otherwise invariant across vertebrate species. Frogs, chickens, rodents, cattle, and nonhuman primates all possess an aspartic acid residue at this location. In humans, the amino acid may be either an aspartic acid, which is the predominant residue at this position, or asparagine. The α5 nicotinic subunit is not involved in receptor binding in vivo
(7), and this variant is located in the cytoplasmic loop between transmembrane domains.
Evidence that the amino acid change is functionally relevant is supported by the fact that, in vitro, α4β2α5 nicotinic receptors with the aspartic acid variant (D398) exhibited a greater maximal response to a nicotinic agonist than did α4β2α5 nicotinic receptors with the asparagine amino acid substitution (N398). Because the allele that codes for asparagine is associated with increased risk for developing nicotine dependence, and nicotinic receptors containing the α5 subunit with this amino acid (N398) exhibit reduced function in vitro, reduced function of α4β2α5 nicotinic receptors may lead to an elevated risk for developing nicotine dependence. The observation that decreased nAChR function is associated with increased risk for nicotine dependence is consistent with the observation that individuals who are extensive metabolizers of nicotine (reduced receptor activation per cigarette) are at increased risk for nicotine dependence
(41,
42) . We believe that this combined evidence of high conservation across species and biological change in receptor function supports the amino acid variant in the α5 nicotinic receptor as a causative biological factor that alters the risk of nicotine dependence, although we cannot definitively rule out the other correlated SNPs across the three-gene cluster.
The α4β2α5-containing nicotinic receptors are expressed on dopaminergic neurons in the striatum
(16), where they modulate nicotine-stimulated dopamine release
(15) . In addition, α4β2α5 nicotinic receptors are also found on γ-aminobutyric acid (GABA)-ergic neurons in the striatum and ventral tegmental area
(43) . This region of the brain is associated with the reward pathway, and the neurotransmitter dopamine plays a crucial role in the development of dependence. Individuals with reduced α4β2α5 cholinergic receptor activity may require greater amounts of nicotine to achieve the same activation of the dopaminergic pathway. Alternatively, reduced activity of the receptor complex on GABA-ergic neurons may lead to increased dopaminergic activity in response to nicotine. How the altered receptor activity caused by the
CHRNA5 amino acid change modifies liability to nicotine dependence via the reward system in response to nicotine requires further study.
The “at risk” allele differs dramatically across human populations. It is predominantly seen in populations of European and Middle Eastern descent and is uncommon or nonexistent in populations of African, Asian, or American origin. Interestingly, African Americans have a lower prevalence of nicotine dependence than European Americans
(44,
45), and this may be explained in part by the low prevalence of this risk allele in populations of African descent.
Less information is available regarding the second independent finding marked by the genetic variant rs578776 in this gene cluster. This SNP, rs578776, is located in the 3′ untranslated region of the CHRNA3 gene. The 3′ untranslated regions contain regulatory sequences, and we can speculate that this SNP is a putative functional variant. It is important also to note that there are correlated SNPs with rs578776 in CHRNA5 and CHRNA3, and the localization of the functional alleles may be in either gene. Further experiments are needed to identify the potential functional variants and the biological mechanisms.
In summary, there are at least two independent genetic variants in the CHRNA5-CHRNA3-CHRNB4 gene cluster on chromosome 15 that are highly associated with smoking behaviors, and we have extended our work to identify a potential biological mechanism for one of the findings. This study provides strong evidence that an amino acid change in the α5 nicotinic receptor, which is highly conserved across species, results in a functional change that is associated with a smoker’s risk of transitioning from nondependence (light smoking) to dependence (habitual smoking) on nicotine. This variant is common in populations of European and Middle Eastern descent and increases the risk of developing nicotine dependence, but it is rare in populations of African, American, and Asian descent. Intriguingly, three recent papers demonstrated that this genetic locus also contributes to the risk of developing lung cancer. A second distinct finding in this gene cluster is also seen, although further study is needed to localize the potential functional allele and to determine whether the variant affects the expression or function of the CHRNA5 or CHRNA3 gene. These converging genetic associations and biological data support the importance of CHRNA5 and potentially CHRNA3 in the development of nicotine dependence and highlight the pharmacogenetic response to nicotine that increases the susceptibility to dependence. These findings may help predict response to pharmacologic therapies, such as varenicline and nicotine replacement, for smokers who attempt to quit, and it may shed important light on the biological mechanisms that contribute to lung cancer.