Following any randomized clinical trial, investigators and biostatisticians can always look back on the design decisions made and argue that some should have been made differently. Such retrospective evaluation is necessary and valuable. Identifying limitations to the inferences drawn from any randomized clinical trial can prevent misinterpretation of clinical and research applications by the media, patients, families, clinicians, or others. There are examples in the medical research showing deleterious effects on patient care that persist as long as 15 years resulting from a study with methodological flaws that were slow to be recognized (for example, see reference
1 ). In this discussion we refrain from indicating what conclusions we draw from the data of the CATIE study. It is only appropriate that each clinician, clinical consumer, or medical policy maker draw his or her own interpretation, taking into consideration the limitations we discuss. More important, however, such retrospective evaluation provides guidance for what might be done differently and better in subsequent randomized clinical trials.
CATIE was reported as being funded by the National Institute of Mental Health (NIMH) to compare “the relative effectiveness of second-generation (atypical) antipsychotic drugs as compared with that of older agents” (
2, p. 1209). This issue is of major public health significance. Approximately 2.4 million Americans suffer from schizophrenia, which imposes a severe emotional and financial burden on patients, families, and society. Trials done before CATIE often were very short (4–8 weeks), were underpowered, and included study groups unrepresentative of the patients clinicians are required to treat. Most important, since the cost of atypical antipsychotics is many-fold larger than that of typical agents, the lack of studies addressing that particular comparison was particularly important. The CATIE study was the largest outcome study of schizophrenia to date. It had multiple levels of input and feedback, resulting in a number of design decisions; some of these helped the quality of its science and the resultant policy implications, and some did not.
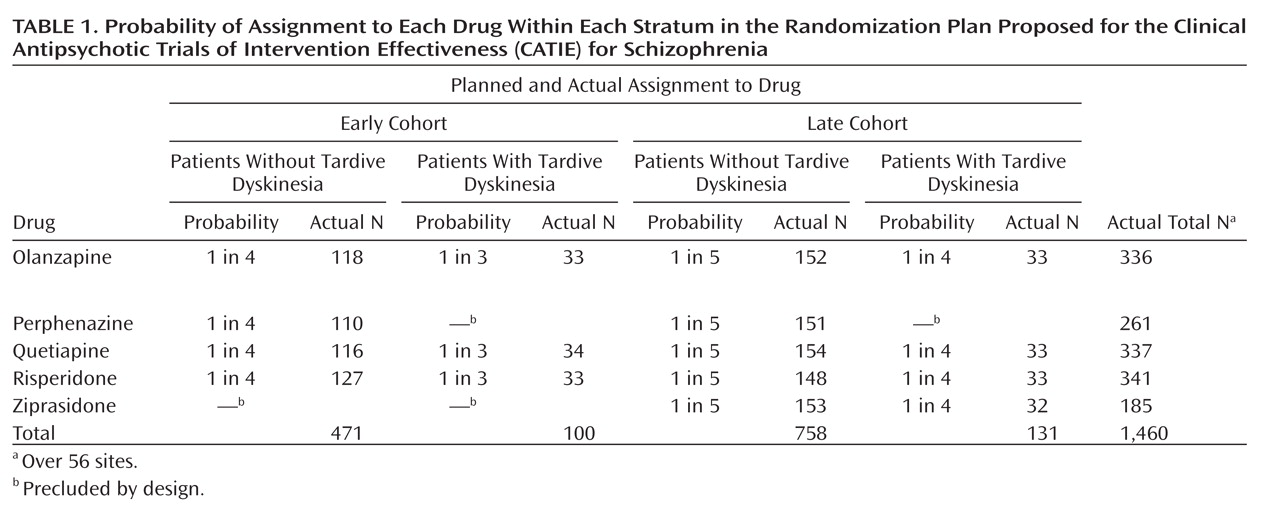
CATIE reports usually state (
2, p. 1209) that 1,493 patients with schizophrenia were recruited at 57 sites and randomly assigned to five drugs—olanzapine, perphenazine, quetiapine, risperidone, and ziprasidone, but to understand CATIE (or any randomized clinical trial), it is necessary to understand its design more precisely. The study group was stratified by the presence or absence of tardive dyskinesia and by cohort—those enrolled before inclusion of ziprasidone (early) and those after (late)—resulting in four strata. Patients were
not randomly assigned to all five drugs, but to a different selection of drugs within each stratum (see
Table 1 ).
Thus, there were five drugs, four strata, and 57 sites to deal with, with 16 cells in the design for each site in an unbalanced design. Of the 56 included sites, 12 had groups containing 15 or fewer patients. These sites were pooled for CATIE’s analysis by type of facility, e.g., Department of Veterans Affairs. In the sites with more than 15 subjects, the median number of subjects per site was only 28. Two outlier sites had 87 and 60 subjects, respectively. Every site had at least one empty cell. Even at the largest site (N=87), there were only four patients with tardive dyskinesia, two in each of the early and late cohorts, and three empty cells. Thus, although the study group initially seems large, the actual number of subjects—given the number of strata, drugs, and sites—was quite small for analyses that require an adequate number of subjects in each cell.
Which Drugs To Be Compared?
The initial motivation for CATIE was reported as arising from the need to compare the newer, more expensive antipsychotics against the older, less costly ones. However, the design of the study suggests the desire of NIMH and the investigators to answer clinical questions about the differential effectiveness of the atypical antipsychotics as well as their possible superiority, individually or as a group, to the typical neuroleptics. Advertising claims for the advantages of each atypical were thought to be influencing practice, but there were few independent direct comparisons of any of the drugs, since the Food and Drug Administration does not require demonstration that new drugs are superior to already available drugs for marketing. Consequently, intensive marketing of newer drugs can, in the absence of an evidence base, mislead clinical decision making. Large, publicly funded studies are crucial to answer questions about the relative benefits and safety of these drugs.
There are always many clinical and practical considerations that affect trial design. In CATIE, not all of the dozen or so available typicals could be assessed. Perphenazine was chosen not because it is the most commonly used or representative of typicals, but because the CATIE investigators thought it could be used effectively at modest doses with fewer extrapyramidal side effects than is the case with haloperidol, the most common typical antipsychotic
(2) . Moreover, investigators were advised by consumer groups that haloperidol should not be used, because patients would be reluctant to enter the trial. Patients with preexisting tardive dyskinesia ethically could not be exposed to any typical neuroleptic. At this point, the goal of comparing typicals versus atypicals clashes with the goal of comparing atypicals against each other. For the first goal, patients with tardive dyskinesia must ethically be excluded. Yet for the second goal, to exclude those with tardive dyskinesia would compromise the intention to make the study results applicable to the broad range of patients seen in actual clinical practice.
CATIE’s decision to include people with tardive dyskinesia but to exclude them from randomization to perphenazine resulted in the stratification by tardive dyskinesia seen in
Table 1 . This stratification accounts for most of the cells with very small numbers, particularly empty cells at individual sites.
Ziprasidone was introduced after the study began. NIMH and the investigators believed it important to include it as well, even though almost one-half of the subjects in CATIE had already been randomly assigned to treatments (
Table 1 ). Choosing to do so, however, introduced further stratification (by cohort) and further reduced the number of subjects in each cell. While olanzapine, quetiapine, and risperidone each had a total of 336–341 patients, perphenazine had only 261 and ziprasidone had fewer yet, 185, with the numbers of subjects in the 16 cells ranging from 33 to 154.
It is almost always possible to design a study optimally to test one hypothesis (“Is treatment 1 better or worse than treatment 2?”), since all design decisions can then be focused on only that one research question. Addition of even one more research question often necessitates design compromises that weaken the answers to both. Had CATIE focused on comparing olanzapine versus perphenazine, for example, the patients with tardive dyskinesia would have been excluded. If it had compared only quetiapine versus ziprasidone, the study would not have started recruitment until ziprasidone was available and need not have stratified for tardive dyskinesia. If it had compared only olanzapine and risperidone, stratification for neither tardive dyskinesia nor cohort would have been necessary.
With two drugs, there is only one comparative pairwise decision to be made. CATIE selected as its threshold of clinical significance (
2, p. 1211) a 12% difference in failure rates. In a single-site, nonstratified study comparing the failure rates of two treatments, 270 patients per treatment are needed to have at least 80% power of detecting effect sizes exceeding that threshold with a two-tailed 5% test
(3,
4) . With stratification by site, tardive dyskinesia, and cohort, with comparisons of five drugs, and with an unbalanced design, the necessary number of subjects must be much larger. Thus, adequate power is possible in CATIE, although not assured, only for comparisons among olanzapine, quetiapine, and risperidone, not for perphenazine (tardive dyskinesia exclusion) and not for ziprasidone (late addition) (
Table 1 ). With five comparative pairwise decisions and no stratification, one would need about 400 patients per drug group. However, in CATIE there were five drugs, hence 10 pairwise decisions, as well as stratification by tardive dyskinesia, cohort, and site.
It is well known that using statistical tests to compare multiple pairs of drugs without adjustment for multiple testing results in a proliferation of false positives. With adjustment for multiple testing without an increase in sample size, power is sacrificed, resulting in a proliferation of false negatives. Even if both false positive and false negative error rates are dealt with in design, the answers may still be ambiguous.
This is not an argument for focusing each randomized clinical trial on comparison of only two drugs. The crucial message is that each study should ask no more research questions than the study can
optimally be designed to answer. For example, if the patients with tardive dyskinesia had been excluded from CATIE and ziprasidone had not been belatedly added, the study could have been a simple, balanced four-treatment comparison with no stratification, such as that in an earlier NIMH-funded multisite randomized clinical trial, the Multimodal Treatment Study of Children With ADHD (MTA)
(5) . In the MTA, in contrast to CATIE, there were fewer sites and they were carefully selected (to avoid post hoc removal of patients or sites and to protect against empty cells). Thus, there was a balanced study group with approximately equal numbers of patients per site, with equal random assignment within each site to the four treatment groups. While such a design in CATIE would not have addressed any research questions for patients with tardive dyskinesia or about ziprasidone, neither does inclusion of the inadequate number of patients in the groups indicated in
Table 1 .
In any research area, there are always numerous research questions of importance and interest. Postponing any is difficult. However, the choice is between answering a few important questions well by optimizing research design decisions for those questions versus addressing many questions inadequately—wasting resources and time and, most important, risking misleading clinical decisions and future research. In the future, the necessity for focusing the number of research questions in any one randomized clinical trial needs to be better appreciated by investigators, reviewers, and sponsors.
A Superiority Study Rather Than an Equivalence Study?
CATIE was designed as a superiority study (to show the superiority of one drug over another), not an equivalence study (to show the clinical equivalence of drugs). That is clear because its results are described in terms of “statistically significant differences,” “p values,” and “power,” all the language of a superiority study.
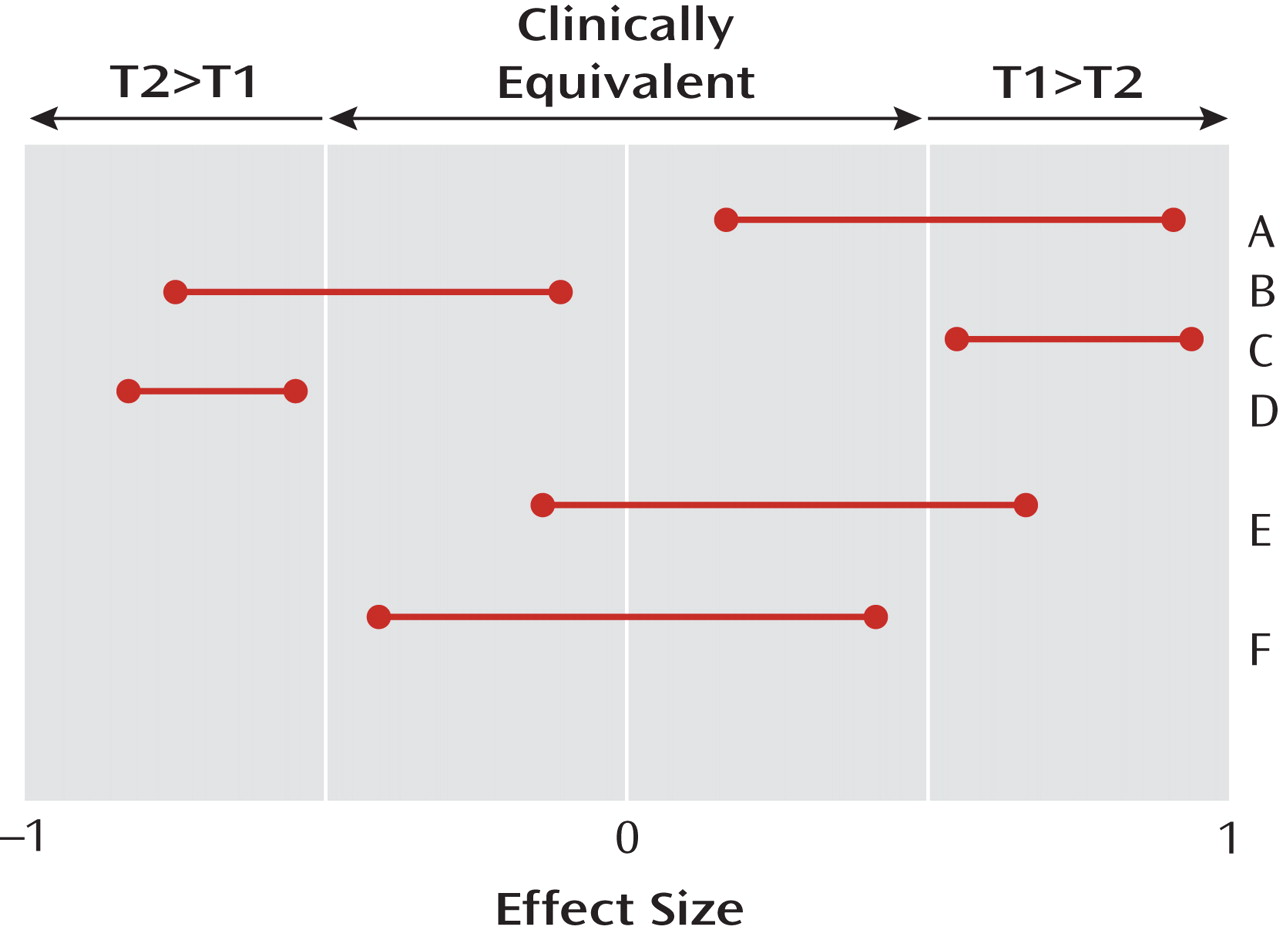
Figure 1 illustrates the difference. The horizontal axis indicates the unknown true treatment effect, which has a value of zero if the two treatments being compared are equivalent (never completely true [
6,
7 ]) and a value of ±1 if every single patient responds better to one of the two drugs (also never completely true). The “number needed to treat” equals the reciprocal of this effect size. The 95% two-tailed confidence intervals for effect sizes comparing different pairs (A, B, C, D, E, F) of hypothetical treatments are displayed. Clinical equivalence exists in a region researchers are required to specify a priori around zero, with values outside that region indicating that one of the two treatments being compared is clinically significantly better than the other (T1>T2 or T2>T1).
A successful
superiority study results in a 95% two-tailed confidence interval for the effect size comparing T1 versus T2 that does not contain the null effect of zero (e.g., A, B, C, and D in
Figure 1 ). If that happens, the result is described as “statistically significant at the two-tailed 5% level.” A study is well designed as a superiority study if, whenever the true unknown effect size exceeds the threshold of clinical significance, the probability of a successful superiority study is greater than, say, 80% (power). A statistically nonsignificant result in such a study (absence of proof) does not mean equivalence (proof of absence) but, usually, inadequate power (e.g., study E in
Figure 1 ). A statistically nonsignificant result in a superiority study should be regarded as a “hung jury” from which, in the absence of other assurances, no conclusions about the treatment effects can be drawn.
In contrast, a successful
equivalence study produces a 95% confidence interval for the effect size that lies completely within the clinically equivalent region (F in
Figure 1 ). A study is well designed to be an equivalence study if, when the true effect size is null, the probability of a successful equivalence study (i.e., both the upper and lower bounds of the effect size confidence interval lie within the clinically equivalent region) is greater than, say, 80%. It should be noted that a clinically equivalent result may be statistically significant or not, that a statistically significant result may be clinically equivalent or not, and that a poorly designed study is likely to result in a result like that in E, neither statistically significant (since the null effect is included) nor equivalent (since clinically significant effects are also included).
Successful equivalence studies typically require much larger samples than do superiority studies. For example, for a simple t test comparing two groups, if the sample size per group necessary for adequate power in a superiority study were 270 (comparable to that needed in CATIE), the necessary sample size for an equivalence study would be 362. With stratification, an unbalanced design, and multiple outcomes, clearly no group size in CATIE approached the level necessary for an adequately designed equivalence study.
This means that any results reported as “not statistically significant” in the CATIE study, particularly comparisons involving perphenazine or ziprasidone, are likely the consequence of a lack of power and should not be interpreted as evidence supporting equivalence. No confidence intervals for pairwise clinically interpretable effect sizes were presented in order to document equivalence. Thus, none of the “not statistically significant” results can be accepted as an indication of equivalence of treatment effects
(8) .
While CATIE reported that olanzapine
was statistically significantly superior to quetiapine and risperidone (
2, p. 1212), many interpretations of CATIE results focus inappropriately on results and secondary outcomes that were not statistically significant
(8) . Despite the fact that the only significant finding from CATIE demonstrated the superiority of olanzapine to quetiapine and risperidone and that the nonsignificant findings related to perphenazine are likely due to inadequate power, as of 2008 it has been reported that the use of olanzapine has decreased and use of perphenazine has increased
(9) .
Nevertheless, the decision to design CATIE as a superiority study was probably wise, given the motivation for the study. However, reporting confidence intervals for a clinically interpretable effect size for each pair of treatments would have given more information than merely reporting statistical significance
(10) and could have established either superiority or equivalence or indicated inadequate power. Future randomized clinical trials should report such confidence intervals rather than merely p values
(10 –
14) . In any case, it is important in planning future studies to clearly articulate the goal of establishing superiority or equivalence, then to plan and execute the study so as to successfully meet that goal, and then to interpret results in a manner consistent with the goal and results.
The Population?
CATIE designated as the population to be sampled at each site patients with a diagnosis of schizophrenia, between 18 and 65 years of age, without major contraindication to any of the drugs to which they were to be randomly assigned (necessary for ethical reasons), and with more than one episode.
Such a choice contrasts with pharmaceutical company studies, which often impose inclusion and exclusion criteria that are severely limiting. They thereby exclude a portion of patients that clinicians are often called upon to treat
(15,
16) and then produce results unlikely to generalize to clinical practice. The patients CATIE chose to study were thought to be more likely to yield results fostering good general clinical decision making.
A Multisite Randomized Clinical Trial?
Another wise CATIE design decision was to structure the randomized clinical trial as a multisite study. In general, there are two major advantages to a multisite study: 1) it is often the only way to generate a large enough sample for adequate power in a superiority study and 2) it allows for testing the generalizability of the conclusion at least across sites like those included in the study (the site-by-treatment interaction). Both reasons apply to CATIE.
A multisite randomized clinical trial is necessarily stratified by site
(17,
18) . The local groups recruited at the various sites often differ from each other. The treatments are delivered in different ways by different research staffs, some more successful in retaining and treating patients than others. Both site effects and site-by-treatment interactions are likely to exist in any multisite randomized clinical trial. If these effects exist and are not considered in the analysis, these effects are remapped to bias the estimated treatment effect (frequently) and to increase error (always), thus increasing the risk of both false positives and false negatives
(18) . Thus, in the MTA study
(5), with major efforts to ensure fidelity to a central protocol, site differences were highly significant but treatment-by-site interactions were not. On the other hand, the Infant Health and Development Program, another multisite study
(19), demonstrated not only highly significant site differences but, despite major efforts to ensure fidelity to a central protocol, also site-by-treatment interactions.
To consider site and site-by-treatment interactions requires that each site have more than a minimal number of subjects in each cell of the design. Because every site in CATIE had empty cells, it was not possible for analysts to fully consider site or to consider the site-by-treatment interaction at all. In future multisite randomized clinical trials (as is often already the case), one criterion for eligibility of a site should be an adequate number of subjects to replicate the full design of the study. In CATIE, that would have required a design with fewer drugs, less stratification, inclusion only of sites with access to a large enough number of patients, or some combination of these.
Further Stratification?
Stratification by site was necessitated by the decision to do a multisite study; stratification by tardive dyskinesia was necessitated by inclusion of perphenazine (not used for tardive dyskinesia patients); stratification by cohort was necessitated by inclusion of ziprasidone (not used in the early cohort). Clearly, this stratification is already troublesome in its effect on both power and precision. Yet some have suggested that CATIE should have been even further stratified, for example, by whether random assignment resulted in a drug switch or not, or by gender, ethnicity, age, etc.
Following is a brief summary of what is known about stratification in randomized clinical trials in general. If such stratification is done when unnecessary, there will likely be a loss of precision and power; if such stratification is not done when necessary, there will also likely be a loss of precision and power. What determines the necessity for stratification is whether some baseline variable moderates the effect of treatment
(20,
21), i.e., whether the effect size comparing two treatments differs depending on the value of the moderator variable. However, if stratification is done on such a moderator, 1) there must be adequate sample size to deal with the stratification in analysis and 2) the interaction between the moderator and choice of treatment must be included in the analysis.
When CATIE was designed, there was no empirical justification for any moderators of treatment response, and CATIE correctly took the course of not stratifying on any variable other than those already necessitated by the design. However, post hoc analysis of CATIE
(22) provided empirical justification for the belief that whether or not randomization resulted in switching drugs may be a moderator of treatment outcome. Thus, while the designers of CATIE were wise in not stratifying on that factor, designers of future randomized clinical trials in this area might be wise to do so. CATIE also did not stratify on gender, age, ethnicity, etc.; again, this was a wise choice in the absence of any empirical justification for doing so at the time of CATIE design. As yet there is still no evidence that such factors moderate treatment effects on this outcome in this chronically ill population.
Researchers and reviewers often demand that the sample be stratified on baseline measures not hypothesized to be moderators of treatment outcome or that such baseline measures be otherwise “controlled” or “adjusted” in a randomized clinical trial, apparently unaware that to do so can carry a heavy cost. For example, if a random sample were selected (about 70% male) from the CATIE population and randomly assigned to the two treatments, the effect size estimated and tested in comparing these groups would be the overall effect size in that population. However, if the true effect size were greater for males than for females (if gender were a moderator of treatment), that overall effect size is technically correct for the population as a whole but still might mislead individual clinical decision making, by underestimating the effectiveness for males and overestimating it for females.
If gender were a moderator and, instead, a sample were obtained that was stratified by gender (to include, say, 50% men) and the analysis included gender, treatment, and their interaction in the model, properly centered
(23), the interaction effect would reflect the difference between the two gender effect sizes and the treatment effect would be the average of the two. If there were a statistically significant interaction, the effect sizes for males and females would then correctly be reported separately. The main effect of the treatment here is the average of the two effect sizes, which would again mislead clinical decision making.
However, if that analysis were done without inclusion of the interaction term, the treatment effect might estimate neither the overall treatment effect in the population nor the average of the two gender effect sizes, but some weighting of the two gender effect sizes depending on the balance of the design and other such factors. In general, when the sample is stratified (or covariates included), the treatment effect cannot be interpreted for clinical decision making, for it depends on which factors are considered, how they are balanced in the design, whether they are correlated with each other, whether they interact with one another and with treatment choice, and how they are included in the analysis. In short, stratification in the design and/or adjustment in the analysis should not be casually done, but only when there is theoretical rationale and empirical justification from previous research to justify such action. Then the study should be designed and analyzed to detect interaction effects.
Choice of Primary Outcome?
Statisticians have long argued for a single primary outcome in a randomized clinical trial. With multiple outcomes, as was the case with multiple drug comparisons, in the absence of adjustment for multiple testing, false positives proliferate. Even with appropriate adjustment and increased sample size, which of the positive results are false positives and which of the negative results are false negatives is unknown.
However, the problems are even more serious with multiple outcomes, for while no patient is in more than one treatment group in a randomized clinical trial, each patient will experience multiple outcomes, simultaneously some benefits and some harms. In addition to its primary outcome measure, CATIE reported six specific measures of effectiveness, many tested both overall and pairwise, and 41 measures of safety, with no adjustment for such multiple testing.
Whether an individual patient is benefited or harmed depends crucially on the particular configuration of harms and benefit that individual experiences and whether the configuration of benefits clinically outweighs the configuration of harms or vice versa. Reporting multiple benefits and multiple harms separately conveys no information on whether these occur in the same or different patients or how the harms balance against the benefits within individual patients. Thus, with multiple outcomes reported separately, it is often impossible to decide which of two treatments is preferable for individual patients.
CATIE, however, proposed a single primary integrative outcome measure, a decision that would have avoided the considerable problems of multiple outcome testing. As in all trials, the decision of which drug is reported as superior in CATIE should be based on the investigators’ a priori choice of primary outcome. The descriptive statistics on secondary outcomes should not be used to modify that decision but are important to provide insight as to how that decision came about. Post hoc “cherry picking” among multiple outcomes raises the risk of misleading results and is a major reason for requiring registration of randomized clinical trials
(24,
25) . With multiple testing and cherry picking, results become at best ambiguous.
CATIE defined an “integrative clinical outcome” as a measure that “integrates patients’ and clinicians’ judgments of efficacy, safety, and tolerability into a global measure of effectiveness that reflects their evaluation of therapeutic benefits in relation to undesirable effects” (
2, p. 1211). However, in implementing the particular outcome measure, CATIE used not time to failure of a drug but, instead, time to discontinuation for any cause. In the absence of such a central protocol governing discontinuations at each site, it was left to the site physicians to make discontinuation decisions. This contributed both to site effects and site-by-treatment interactions.
Approximately one-half of the discontinuations were “owing to patient’s decision” rather than specifically to failure of the drug, and such discontinuations may not reflect drug failure at all, but, for example, dissatisfaction with study participation. Also, in CATIE, patients were informed that they could discontinue at any time and be switched to another drug in phases II and III. Since no drug currently can reasonably be expected to cure schizophrenia, this offer may have encouraged patients, families, or physicians to give up on a drug prematurely in hopes of something better
(26) .
In CATIE, this problem could even now be mitigated by survival analyses, treating discontinuation due to treatment failure as an outcome and discontinuation for any other reason as a censored data point. However, that would profoundly change the results. For example, CATIE has been reported to show about a 70% discontinuation rate for these antipsychotics, but the actual failure rate may be half that. Future studies might follow CATIE’s lead in proposing a single primary integrative outcome but focusing only on treatment failure for any reason and defining exactly what “treatment failure” means for all the sites. Finally, conclusions reached by using the selected primary outcome measure might determine the recommendations of the study, with secondary outcomes only illuminating what that recommendation might mean.
Conclusions
CATIE investigators may have attempted to do the impossible: to compare the atypicals not only to each other but also to at least one typical antipsychotic, to add a new antipsychotic halfway through the study, and to do so in a heterogeneous study group without defining or constricting clinical decision making in any way. A consistent theme in this evaluation of CATIE is too many drugs, too many strata, too many sites with inadequate numbers of subjects, too many outcome measures considered separately in drawing conclusions, and too many statistical tests. Each additional drug, stratum, etc., increases the sample size per site necessary for adequate power in valid analyses. While a study group of 1,493 initially seems large, the design decisions eroded that to a study group too small to detect either superiority (arguably other than that for olanzapine over quetiapine and risperidone) or equivalence effects. The lesson is that sponsors and grant reviewers need to be as cautious and proactive about power as the investigators.
CATIE provides valuable lessons for the design of future studies. Both sponsors and investigators are appropriately anxious to “get their money’s worth.” Indeed, every effort must be made to use the resources available to answer as many important and interesting questions as can be answered optimally (not necessarily perfectly), for the conclusions must effectively guide future research efforts as well as clinical decision making. However, no one study can answer all the important, interesting research questions. Attempting to do so compromises any attempt to definitively answer any research question.