An unexpected hint from a rat model of psychosis led scientists to propose a new strategy to treat schizophrenia. In 1998, Bita Moghaddam and Barbara W. Adams
(1) observed that activation of group II metabotropic glutamate receptors (mGluRs) by LY354740 (an Eli Lilly agonist of mGluR2/mGluR3) in rats injected with phencyclidine attenuated the disruptive effects of phencyclidine on working memory, stereotypy, locomotion, and cortical glutamate efflux. This behavioral reversal occurred in spite of sustained dopamine hyperactivity. The finding opened a new approach to research on the potential treatments of psychiatric disorders that has extended the range of possible therapeutic targets beyond the dopamine hypothesis of schizophrenia. Almost a decade later, this discovery was followed by a clinical report
(2) showing that agonists of group II mGluRs (LY404039 administered as the prodrug LY2140023) improved positive symptoms in patients with schizophrenia, with effects similar in magnitude to those of standard olanzapine treatment.
Glutamate, the major excitatory neurotransmitter in the brain, exerts fast effects by regulating ion flow, through AMPA,
N -methyl-
d -aspartic acid (NMDA), and kainate receptors, and modulatory effects by acting on different subtypes of G-protein coupled mGluRs. Converging lines of evidence implicate altered glutamatergic neurotransmission in the pathophysiology of schizophrenia
(3) . For example, exposure to NMDA receptor antagonists, such as phencyclidine or ketamine, produces “schizophrenia-like” symptoms in healthy individuals and exacerbates pre-existing symptoms in patients with schizophrenia
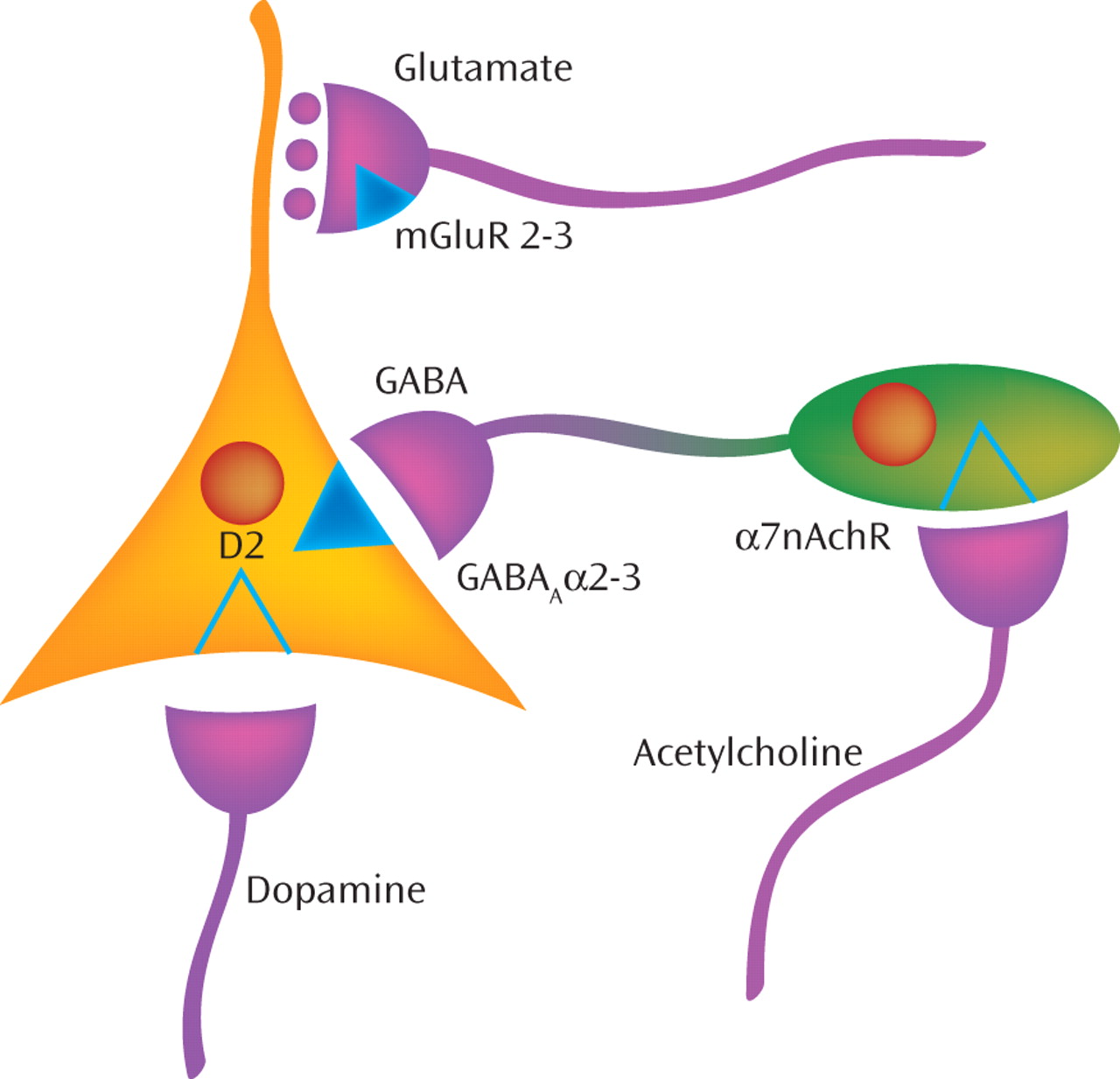
(4) . Such NMDA receptor antagonists are thought to mimic an intrinsic “hypofunction” of NMDA receptors on inhibitory γ-aminobutyric acid (GABA) neurons in schizophrenia, which leads to reduced inhibition of excitatory neurons and a consequent deleterious excess of glutamate release that disrupts the coordinated activity of neuronal networks (
5,
Figure 1 ]). Because activation of the mGluR2 subtype of mGluRs, which is located on the terminals of excitatory axons, suppresses glutamate release, mGluR2 agonists were predicted in a clinical trial
(6) to be beneficial in counteracting the consequences of the hypothesized “NMDA receptor hypofunction” in schizophrenia. Consistent with this prediction, the clinical trial provided proof-of-concept evidence that a novel compound with agonist activity at both mGluR2 and mGluR3 receptors had antipsychotic efficacy in schizophrenia
(2) .
Although these initial findings are quite exciting, they raise a number of questions that are important for the continued development of novel antipsychotic medications with this mechanism of action. For example, what is the status of the targeted receptors in schizophrenia? Which regions of the brain contain altered levels of the receptors? Does the nature of the alterations in mGluR2 and mGluR3 receptors in schizophrenia suggest that the apparent antipsychotic efficacy of mGluR2/ mGluR3 agonists is principally at one of these receptor subtypes? Until recently, studies were carried out with orthosteric agonists, which activate both receptor subtypes (mGluR2 and mGluR3) with equal affinity. However, mGluR2 and mGluR3 are desensitized differently
(10), which raises the possibility of different roles for these two subtypes of group II mGluRs in the treatment development of psychiatric disorders.
In order to address these types of questions, Ghose and colleagues in this issue of the
Journal (7) measure the protein levels of mGluR2 and mGluR3 receptors in postmortem tissue samples of three cortical regions in subjects with schizophrenia and matched normal comparison subjects. The investigators also measure protein levels of the enzyme glutamate carboxypeptidase II (GCP II), which metabolizes
N -acetylaspartylglutamate (NAAG), an endogenous agonist of mGluR3
(11) . Relative to comparison subjects, individuals with schizophrenia had significantly higher levels of GCP II protein and lower levels of mGluR3 protein (with no difference in mGluR2 protein levels) in the dorsolateral prefrontal cortex. In contrast, no differences between subject groups were found for any of the three proteins in the temporal or motor cortex. The authors conclude that these findings implicate the mGluR3 receptor as the principal mediator of the antipsychotic action of mGluR2/mGluR3 agonists.
One approach to interpreting the meaning of these types of protein alterations involves determining how each alteration fits into the cascade of molecular features that characterize the underlying disease process of schizophrenia. That is, any given finding in a postmortem study could represent a cause (an upstream factor related to the pathogenesis of the illness), a consequence (a deleterious effect of a cause), a compensation (a response to either a cause or consequence that helps restore homeostasis in the brain), or a confound (a product of factors frequently associated with, but not a part of, the disease process or an artifact of the approach used to obtain the measure of interest)
(12) . Ghose and colleagues have carefully examined potential confounds in their study, and the available evidence excludes the usual suspects of between-group differences in subject characteristics (e.g., age and sex) or tissue quality (e.g., delay between time of death and tissue preservation) or of the effect of chronic exposure to antipsychotic medications. For example, the authors show that levels of mGluR3 and GCP II proteins, which have similar distributions in human and rodent brain, were not altered in rats exposed for 6 months to antipsychotic drugs.
As for any empirical data, the strongest argument that a given finding reflects the disease process is the independent replication of the findings using complementary research strategies that tend to be subject to a different set of confounds (e.g., postmortem studies of GCP II and mGluR3 mRNA levels or in vivo measures of mGluR3 binding). A recent report by Gonzalez-Maeso and colleagues
(13) demonstrated changes in mRNA expression of mGluR2-5-hydroxytryptamine 2A receptor (2AR) in psychosis, not mGluR3. In the postmortem human brain, mGluR2 forms a protein-protein functional complex between serotonin and glutamate receptors (2AR-mGluR2). Thus, we cannot exclude the possibility that both receptors are involved in the development or potential treatment of schizophrenia.
An interesting question is regarding the potential relationship between the reciprocal alterations in GCP II and mGluR3 protein levels. Do they represent cause and consequence or consequence and compensation? For example, if the lower levels of mGluR3 protein reflect the effect of risk variants in the
GRM3 gene
(14), then one might predict a compensatory response of lower levels of GCP II in order to increase levels of the endogenous mGluR3 agonist, NAAG, and thus augment stimulation of the apparent deficit in the number of mGluR3 receptors. Contrary to this prediction, the findings of Ghose and colleagues raise the possibility of two separate processes resulting in deficiencies in both an endogenous agonist for the mGluR3 receptor and the receptor itself that converge to produce a marked impairment in this signaling pathway in the dorsolateral prefrontal cortex of individuals with schizophrenia. The combined deficit could thus be considered to support the authors’ interpretation that augmenting activity at the mGluR3 receptor accounts for the reported antipsychotic efficacy action of a mGluR2/mGluR3 agonist in schizophrenia
(2) . Evidence that the combined deficit is present in brain regions typically associated with psychosis (e.g., the striatum and superior temporal gyrus) would strengthen this interpretation. The presence of these alterations selectively in the dorsolateral prefrontal cortex raises the possibility that mGluR3 agonists might improve cognition in schizophrenia.
On the other hand, the combination of less endogenous ligands and fewer receptors raises the possibility that down-regulating mGluR3 signaling is a compensatory response to some other deleterious effect on brain function in the illness. Although the present findings do not immediately suggest a cause and compensation relationship, this interpretation might be consistent with findings in mice indicating that activation of mGluR2, but not mGluR3, receptors explains the reported antipsychotic efficacy of an mGluR2/mGluR3 agonist
(15) . Of course, multiple other proteins are also involved in regulating glutamate neurotransmission in the brain. A fuller knowledge of how the profiles of these proteins are altered across individuals with schizophrenia and across brain regions within individuals might both inform the relationships between such alterations and strengthen predictions regarding which molecules are most likely to be promising targets for novel pharmacological interventions.