Atypical antipsychotic drugs generally share more potent antagonism for 5-HT
2A than dopamine D
2 receptors (
1,
2). However, there are significant differences among these agents in their relative affinities for 5-HT
1A, 5-HT
2C, 5-HT
7, alpha-adrenergic, histamine H
1, muscarinic, and other receptors that may affect their efficacy and tolerability (
2). Genetic polymorphisms in receptor proteins, as well as in cytochrome P450 isoenzymes, contribute additional between-drug variability in clinical effect (
3). Thus, atypical antipsychotics do not produce uniform clinical responses in all patients, and it remains important to have multiple antipsychotic drug treatment choices to address unmet therapeutic needs in patients with schizophrenia and other psychotic disorders (
4,
5).
Lurasidone is a novel psychotropic agent that has been shown in studies of cloned human receptors to be an antagonist at the 5-HT
2A receptor, with a binding affinity (Ki; the dissociation constant of the inhibitor) of 0.47, and a Ki of 0.99 at the D2 receptor. It also has a very high affinity for the 5-HT
7 receptor (Ki, 0.49), which is nearly identical to its affinity for the 5-HT
2A receptor. In addition, lurasidone has moderate partial agonist effects at the 5-HT
1A receptor (Ki, 6.4) and moderately potent antagonist effects at α
2c receptor subtypes (Ki, 10.8) (
6).
The primary objective of this phase 3 study was to evaluate the efficacy of two dosages of lurasidone (40 and 120 mg/day) compared with placebo in the treatment of patients suffering from an acute exacerbation of chronic schizophrenia. The key secondary objective was to evaluate the efficacy of lurasidone compared with placebo in improving the Clinical Global Impressions severity (CGI-S) score. Another major secondary objective was to evaluate the safety and tolerability of the 40 mg and 120 mg doses of lurasidone during 6 weeks of treatment.
Method
This was a prospective, multicenter, parallel-group study in which recently admitted acutely ill inpatients with schizophrenia with an acute exacerbation of psychotic symptoms were randomly assigned to receive 6 weeks of double-blind treatment with once-daily doses of 40 mg or 120 mg of lurasidone, 15 mg of olanzapine (included to establish assay sensitivity), or placebo. The study was conducted between January 31, 2008, and June 16, 2009, enrolling a total of 478 patients at 25 sites in the United States (N=286), five in Colombia (N=48), four in Lithuania (N=29), and 18 in Asia (India, 14 sites [N=89]; Philippines, four sites [N=26]).
All patients who entered the trial reviewed and signed an informed consent document explaining study procedures and potential risks before study entry. The study protocol and all related forms and amendments were approved by an independent ethics committee associated with each study center. The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practices guidelines and with the ethical principles of the Declaration of Helsinki. An independent data and safety monitoring board reviewed unblinded safety and clinical outcome data.
Entry Criteria
Hospitalized male and female patients 18–75 years of age who met DSM-IV criteria for a primary diagnosis of schizophrenia as determined by the Mini International Neuropsychiatric Interview (
8) were enrolled. Patients were also required to have an illness duration of at least 1 year and to have been hospitalized for ≤2 weeks for an acute exacerbation of psychotic symptoms and, at the screening and baseline visits, to have a CGI-S score ≥4 (moderate or greater) and a Positive and Negative Syndrome Scale (PANSS) total score ≥80, including a score ≥4 (moderate) on two or more of the following PANSS items: delusions, conceptual disorganization, hallucinations, unusual thought content, and suspiciousness.
Study Medication
All study medication was identically overencapsulated to preserve the double-blind. A unique participant number was assigned by interactive voice response system when a patient entered the screening phase. At baseline (day 0), patients who continued to meet all study inclusion criteria were randomly assigned via interactive voice response system (in a 1:1:1:1 ratio) to one of four treatment arms: lurasidone, 40 mg; lurasidone, 120 mg; olanzapine, 15 mg; or placebo. Study medication was administered in the morning with a meal or within 30 minutes after eating. Participants assigned to receive lurasidone started treatment at their target dose; patients assigned to olanzapine treatment received 10 mg on days 1–7 and 15 mg thereafter. The olanzapine dosage of 15 mg/day was selected because it is widely used and because there is substantial evidence that it is an effective dosage in patients with schizophrenia, with little evidence that higher dosages offer additional efficacy advantages (
9,
10). This dosage is also consistent with the olanzapine package insert (
http://pi.lilly.com/us/zyprexa-pi.pdf), which states that efficacy in schizophrenia has been demonstrated in a dosage range of 10–15 mg/day, with higher doses not demonstrated to be more efficacious.
Limited use of benzodiazepines was permitted for severe anxiety, agitation, or insomnia. Participants were eligible for hospital discharge to a stable residence after 21 days of treatment if they had a CGI-S score ≤3.
Assessments
The screening evaluation consisted of the Mini International Neuropsychiatric Interview, medical and psychiatric histories, a physical examination, measurement of vital signs, ECG, and laboratory tests.
Efficacy was assessed using the PANSS total and subscale scores (including a post hoc analysis of a modified version of the cognitive subscale, consisting of items P2, N5, N7, G10, G11) (
11,
12), the CGI-S, and the Montgomery-Åsberg Depression Rating Scale (MADRS;
13). PANSS and CGI-S evaluations were performed at the screening and baseline visits and, during treatment, on day 4 and at each of weeks 1 through 6. The MADRS was administered at the screening and baseline visits and at weeks 3 and 6.
Extrapyramidal symptoms were assessed with the Simpson-Angus Rating Scale (
14), the Barnes Rating Scale for Drug-Induced Akathisia (
15), and the Abnormal Involuntary Movement Scale (
16). Safety evaluations included vital signs, weight, laboratory tests (including fasting lipids, glucose, glycosylated hemoglobin [HbA
1c], and insulin), 12-lead ECG, and reported adverse events. Insulin resistance and beta-cell function were measured using the homeostasis model assessment for insulin resistance (HOMA-IR) method (
17).
Statistical Methods
A power calculation was performed that incorporated Bonferroni's procedure for controlling pairwise differences with placebo and was obtained via computer simulations. Assuming that lurasidone differed from placebo in the change from baseline in PANSS total score by 6.8 and 10.0 for the 40 and 120 mg/day dosages, respectively, and further assuming a standard deviation of 19.1, we calculated that 120 patients per group would provide 97% power (at an alpha level of 0.05, two-sided test) to reject the null hypothesis of no difference between placebo and at least one of the lurasidone dosage groups.
The primary efficacy analysis was performed on the intent-to-treat sample, which consisted of all participants assigned to a treatment group who received at least one dose of study medication, had a baseline PANSS assessment, and had at least one postbaseline PANSS assessment during the 6-week study. The primary efficacy measure was the change from baseline in PANSS total score at week 6, and the key secondary efficacy measure was the change from baseline in CGI-S score at week 6. Both measures were evaluated by a mixed-model repeated-measures analysis with an unstructured covariance matrix. The model included factors for pooled center, time (including all scheduled postbaseline assessment visits as a categorical variable), baseline PANSS total score or CGI-S score, treatment, and treatment-by-time interaction. The p values for the comparison of each lurasidone group with the placebo group at week 6 on change from baseline in PANSS total score and CGI-S score were adjusted for multiple comparisons using the Hommel-based tree-gatekeeping procedure to control the family-wise type I error rate (
18). The olanzapine treatment group, which was included to confirm the assay sensitivity of the study, was compared with placebo using the same mixed-model repeated-measures model, without the multiple comparison adjustment. A post hoc mixed-model repeated-measures analysis of the PANSS total score and CGI-S score was also performed comparing the 40 mg and 120 mg lurasidone treatment groups to the olanzapine treatment group.
A prespecified secondary analysis was conducted for change in PANSS total score and CGI-S score, using an analysis of covariance (ANCOVA) model.
Secondary efficacy measures, including PANSS subscale scores (positive, negative, and general psychopathology) and MADRS total score, were evaluated using similar mixed-model repeated-measures models. A post hoc analysis of the modified PANSS cognitive subscale was also performed. Participants who had an improvement of at least 20% from baseline in PANSS total score at week 6 endpoint (last observation carried forward) were defined as “responders.” A logistic regression was performed using the responder outcome as the dependent variable, treatment as a categorical factor, and baseline PANSS total score as a covariate.
The Cohen's d effect size was calculated for week 6 efficacy measures as the between-treatment difference score divided by the pooled standard deviation. For adverse events, number needed to harm was calculated as 1 divided by the difference in the risk of an adverse event for active drug compared with placebo.
Significance testing of selected safety parameters was performed based on a nonparametric rank ANCOVA with baseline value as a covariate, not adjusted for multiple comparisons.
Results
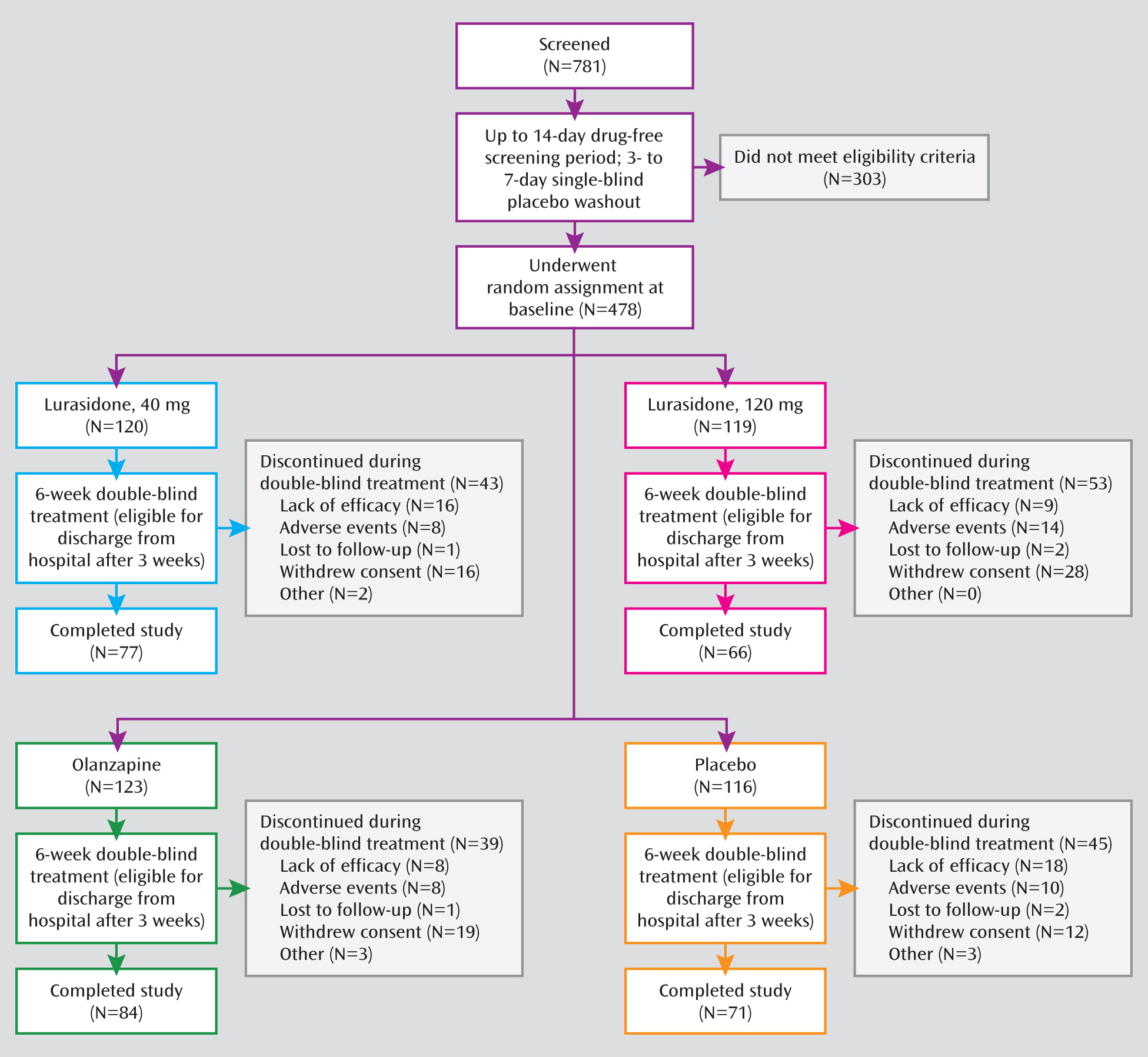
Of a total of 781 patients who were screened and entered the washout period, 478 were randomly assigned to 6 weeks of double-blind treatment (
Figure 1). Baseline demographic and clinical characteristics were comparable among the four treatment groups (
Table 1). The proportion of patients in the lurasidone 40 mg group who completed the study treatment (64.2%) was similar to the proportions who completed treatment in the placebo group (61.2%) and the olanzapine group (68.3%); a somewhat lower proportion of patients in the lurasidone 120 mg group completed the study treatment (55.5%) (
Figure 1).
Efficacy
Based on the mixed-model repeated-measures analysis, the change from baseline to week 6 in PANSS total score was significantly greater for the lurasidone 40 mg (–25.7; adjusted p=0.002) and 120 mg (–23.6; adjusted p=0.022) groups compared with the placebo group (–16.0) (
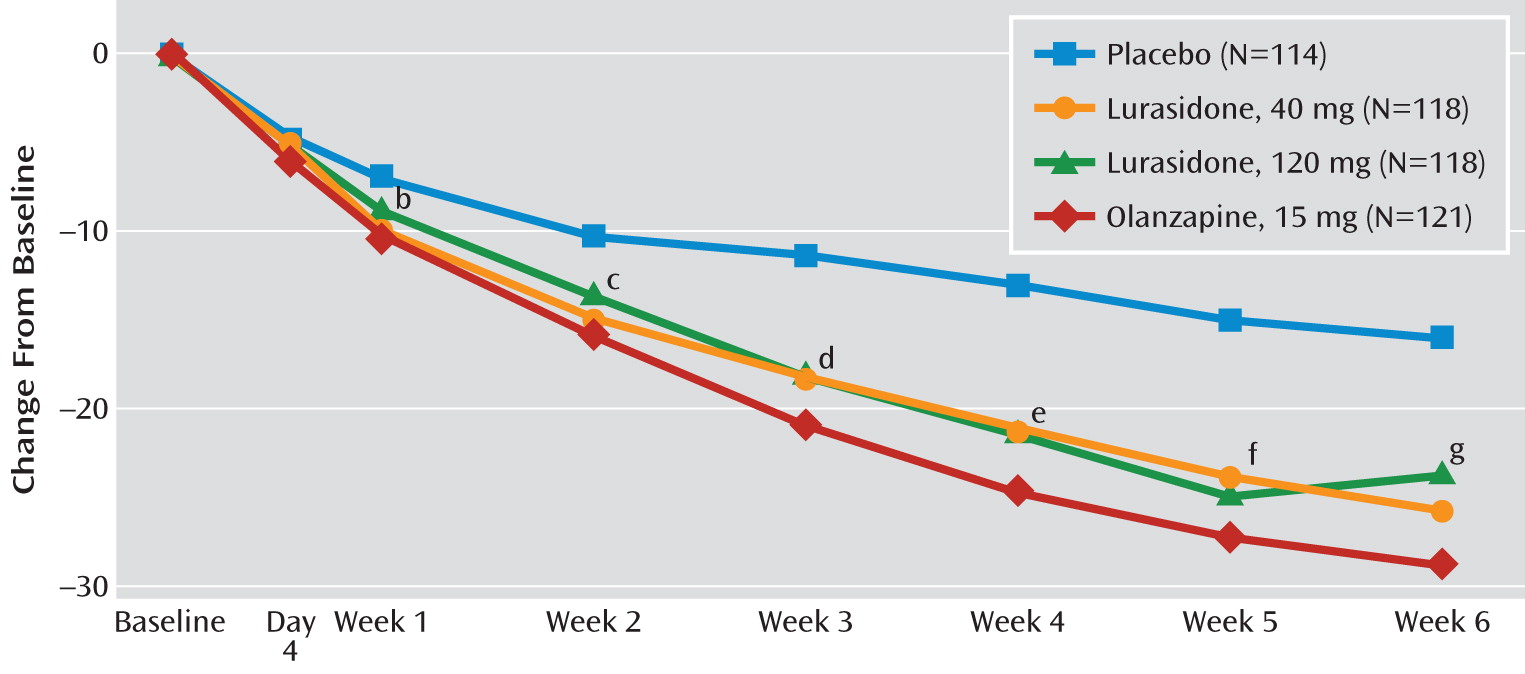
Table 2). The change in PANSS total score was also significantly greater for the olanzapine group (–28.7, p<0.001), thus confirming the assay sensitivity of the study. Statistically significant separation from placebo on the PANSS total score was observed from week 1 onward for the lurasidone 40 mg and olanzapine groups, and from week 3 onward for the lurasidone 120 mg group (
Figure 2; see also Table S1 in the online data supplement). Treatment with both dosages of lurasidone and with olanzapine was also associated with significantly greater improvement at week 6 compared with placebo on the PANSS positive, negative, and general psychopathology subscale scores (
Table 2; see also Table S1 in the online data supplement). Based on a post hoc analysis, treatment with both dosages of lurasidone, as well as with olanzapine, was also associated with significantly greater improvement at week 6 on the modified PANSS cognitive subscale score (see
Table 2).
For the CGI-S score, the change from baseline to week 6 was also significantly greater for the lurasidone 40 mg (–1.5; adjusted p=0.011) and 120 mg (–1.4; adjusted p=0.040) groups compared with the placebo group (–1.1; see
Table 2). The change in CGI-S score was also significantly greater for the olanzapine group (–1.5; p<0.001). Statistically significant separation from placebo on the CGI-S was observed from week 1 onward for the lurasidone 120 mg group, and from week 2 onward for the lurasidone 40 mg group and the olanzapine group compared with the placebo group (see Table S1 and Figure S1 in the online data supplement).
In a post hoc mixed-model repeated-measures analysis of PANSS total score and CGI-S score, there was no statistically significant difference in least-squares mean change scores at week 6 for the olanzapine group compared with either lurasidone group.
In a secondary analysis, an ANCOVA was performed on change from baseline to week 6 (last observation carried forward) for PANSS total score and CGI-S score. In this analysis, the least-squares mean change in PANSS total score was significantly greater for the lurasidone 40 mg (–23.1, p=0.001; effect size, 0.43) and 120 mg (–20.0, p=0.049; effect size, 0.26) groups compared with the placebo group (–15.2). Similarly, the least-squares mean change in PANSS total score was also significantly greater for the olanzapine group (–26.7, p<0.001). In an ANCOVA analysis of CGI-S score, least-squares mean change at week 6 (last observation carried forward) was significantly greater for the lurasidone 40 mg group compared with the placebo group (–1.2, p=0.012), but the comparison with the placebo group was not significant for the lurasidone 120 mg group. The least-squares mean change in CGI-S score was significant for the olanzapine group (–1.4, p<0.001). The results of these sensitivity analyses for PANSS total score and CGI-S score were similar to, and support, the results of the primary mixed-model repeated-measures analysis. Furthermore, on a pairwise comparison, there were no significant differences in endpoint change between the two lurasidone groups on PANSS total score or CGI-S score.
In a logistic regression analysis, responder rates (compared with placebo) and associated odds ratios at 6 weeks (last observation carried forward) were not significant for either of the lurasidone groups, but the comparison was significant for the olanzapine group (a responder rate of 74%, compared with a rate of 49% for placebo; odds ratio=2.9, p<0.001).
Improvement on the MADRS at week 6 was not significantly different between either of the lurasidone groups and the placebo group, whereas the olanzapine group showed significantly greater improvement compared with the placebo group (
Table 2; see also Figure S2 in the online data supplement).
The ANCOVA subgroup analyses showed no significant treatment interactions by gender, race, ethnicity, region, or age for either the PANSS total score or the CGI-S score.
Safety
Adverse events.
A comparable proportion of patients in the lurasidone 40 mg group and in the placebo group reported experiencing at least one adverse event (
Table 3); the incidence was somewhat higher in the lurasidone 120 mg group and the olanzapine group. The majority of adverse events in all treatment groups were rated as mild to moderate. Rates of discontinuations due to adverse events were relatively low in the lurasidone 40 mg group (6.7%), the lurasidone 120 mg group (11.8%), and the olanzapine group (6.5%) and were comparable to those in the placebo group (8.6%). The four adverse events with the highest incidence relative to placebo (that is, the largest drug-versus-placebo difference; see
Table 3) were, for the lurasidone 40 mg group, akathisia (11.8%), agitation (11.8%), nausea (10.9%), and parkinsonism (9.2%); for the lurasidone 120 mg group, akathisia (22.9%), somnolence (15.3%), sedation (13.6%), and parkinsonism (11.0%); and for the olanzapine group, increased weight (20.5%), sedation (14.8%), dry mouth (9.8%), and akathisia (7.4%).
Physical examination and vital signs.
There were no clinically significant treatment-emergent changes in either of the lurasidone groups or the olanzapine group compared with the placebo group in pulse rate, systolic or diastolic blood pressure, or body temperature.
Extrapyramidal symptoms.
The proportion of patients treated with an anticholinergic medication was similar in the lurasidone 40 mg group (20%) and the olanzapine group (18%) but higher in the lurasidone 120 mg group (41%). A smaller proportion of patients in the placebo group (9%) used anticholinergic agents. Benztropine was the most frequently prescribed medication for parkinsonism-related adverse effects (see Table S2 in the online data supplement), followed by trihexyphenidyl, propranolol, and biperiden. The proportions of patients reporting the extrapyramidal symptom-related adverse events of parkinsonism, tremor, dystonia, and akathisia during study treatment are listed in
Table 3. No episodes of opisthotonos were reported in any treatment group. Two patients (1.7%) in the lurasidone 120 mg group discontinued the drug because of extrapyramidal adverse events, whereas none in the other three treatment groups did. The mean endpoint changes in Simpson-Angus Rating Scale score and in the Barnes Rating Scale for Drug-Induced Akathisia global clinical assessment scores were small and not clinically significant in the majority of patients. In the lurasidone 40 mg group and the olanzapine group, change from baseline in the least-squares mean Simpson-Angus and Barnes scale scores was not significantly different from that of the placebo group (see Figures S3 and S4 in the online data supplement). In contrast, the lurasidone 120 mg group had significantly greater Simpson-Angus and Barnes change scores compared with the placebo group. Consistent with these data, the proportions of patients reporting categorical worsening on the Barnes scale, the Abnormal Involuntary Movement Scale, and the Simpson-Angus scale were higher in the lurasidone 120 mg group compared with the lurasidone 40 mg, olanzapine, and placebo groups (see Tables S3–S5 in the online data supplement).
Body weight, body mass index (BMI), and waist circumference.
The mean endpoint changes in weight, BMI, and waist circumference (last observation carried forward) were similar for both lurasidone groups and the placebo group (
Table 4). In contrast, there was a significant mean increase in the olanzapine group compared with the placebo group in both weight (4.1 kg compared with 0.6 kg, p<0.001; see Figure S5 in the online data supplement) and BMI (1.4 units compared with 0.2 units, p<0.001). The proportion of patients with a clinically significant weight gain (≥7% over baseline at last-observation-carried-forward endpoint) was also similar in the lurasidone 40 mg (7.6%) and 120 mg groups (4.2%) compared with the placebo group (7.0%), while the proportion in the olanzapine group was higher (34.4%).
Metabolic parameters.
Endpoint changes in levels of cholesterol, triglycerides, high-density lipoprotein (HDL), and low-density lipoprotein (LDL) (last observation carried forward) were comparable for both lurasidone groups and the placebo group, while the olanzapine group showed a significant mean increase compared with the placebo group in levels of cholesterol (+9.6 mg/dl compared with –6.8 mg/dl, p<0.001), LDL (+4.3 mg/dl compared with –1.8 mg/dl, p=0.010), and triglycerides (+50.0 mg/dl compared with +0.1 mg/dl, p<0.001) (see
Table 4; see also Figure S6 in the online data supplement). Endpoint changes in glucose, HbA
1c, insulin, and HOMA-IR levels were also comparable for both lurasidone groups and the placebo group, while the olanzapine group showed a significantly higher mean increase compared with the placebo group in glucose level (+10.3 mg/dl compared with +0.4 mg/dl, p=0.036) and numerically higher mean increases in levels of HbA
1c (+0.18% compared with –0.05%) (see
Table 4; see also Figure S7 in the online data supplement), insulin (+5.9 mU/liter compared with –2.4 mU/liter), and HOMA-IR (+2.61 U compared with –1.32 U; see Figure S8 in the online data supplement).
Prolactin and other laboratory values.
The median change in prolactin level at endpoint (last observation carried forward) was comparable in the lurasidone 40 mg and placebo groups (+0.7 ng/ml and –0.7 ng/ml, respectively) but was significantly higher compared with the placebo group in the lurasidone 120 mg group (+4.5 ng/ml, p<0.001) and the olanzapine group (+3.8 ng/ml, p<0.001) (see
Table 5). A treatment-emergent shift from normal to high (abnormal) prolactin levels occurred in 23/183 male patients (12.6%; criterion for high value, >17.7 ng/ml) and 10/51 female patients (19.6%; criterion for high value, >29.2 ng/ml) in the combined lurasidone groups, in 8/94 males (8.5%) and 8/27 females (29.6%) in the olanzapine group, and in 6/89 males (6.7%) and 2/25 females (8.0%) in the placebo group. No patient in any treatment group discontinued study medication because of an elevated prolactin level, and treatment with lurasidone had no reported effect on menstrual cyclicity.
ECG.
Treatment with lurasidone was not associated with any treatment-emergent ECG abnormalities compared with placebo. The mean endpoint change in the QTcF interval (last observation carried forward) was similar for the lurasidone 40 mg and 120 mg groups (+5.1 msec and +4.5 msec, respectively), the olanzapine group (+4.4 msec), and the placebo group (+3.8 msec). With the exception of one patient in the lurasidone 40 mg group who had a QTcF >450 msec at baseline that persisted at study endpoint, no QTcF intervals >450 msec were observed in any patient in any of the treatment groups. No patient in any treatment group had an increase ≥60 msec in QTcF interval or a QTcF interval >500 msec.
Discussion
The results of this multicenter, double-blind, placebo-controlled phase 3 trial indicate that lurasidone, at fixed dosages of 40 and 120 mg/day, was an effective and well-tolerated treatment for patients experiencing an acute exacerbation of chronic schizophrenia. Both dosages produced significant improvement on PANSS total score (the primary outcome measure) and CGI-S score (the key secondary outcome measure). There were no significant differences in the efficacy of lurasidone by gender, race, ethnicity, or age. Significant improvement was also observed on the PANSS positive, negative, and general psychopathology subscales and on the modified PANSS cognitive subscale (in a post hoc analysis). The validity of the modified PANSS cognitive subscale is not well established, but our results here are consistent with those from a previous 6-week clinical trial of lurasidone (
7). In addition, in a double-blind trial (
19), treatment with lurasidone was associated with improvement on the Schizophrenia Cognition Rating Scale, an interview-based assessment of cognition-related functioning.
No apparent dose-response relationship was observed in PANSS total score at week 6 in the ANCOVA analysis of the two lurasidone dosages. In fact, the effect size of the change in PANSS total score at week 6 (last observation carried forward) was smaller for the lurasidone 120 mg group than for the 40 mg group (Cohen's d, 0.26 and 0.43, respectively). The reason for the smaller effect size is uncertain, but an inspection of baseline variables (
Table 1) suggests that there were small, but possibly clinically relevant in aggregate, differences in illness severity between the 120 mg and 40 mg groups, namely, a lower age at illness onset (22.7 years and 23.9 years, respectively), a longer duration of illness (14.7 years and 13.3 years, respectively), a higher proportion of patients with ≥4 previous hospitalizations (54% and 43%, respectively), and a higher mean PANSS total score (97.9 and 96.6, respectively). The only previously published trial (
7) that used a fixed dose of lurasidone (80 mg) found an effect size of 0.44, which is comparable to that observed with the 40 mg dose in the present study.
Treatment with the active comparator, olanzapine, was associated with significant endpoint improvement on PANSS total score, CGI-S score, and other secondary measures, including the modified PANSS cognitive subscale. Olanzapine was included to confirm the assay sensitivity of the study; thus, no between-group efficacy testing was planned. However, based on a post hoc mixed-model repeated-measures analysis, no statistically significant difference was observed for change in PANSS total score between either of the lurasidone groups and the olanzapine group. These exploratory findings need to be confirmed in further comparative studies.
The onset of significant improvement in PANSS total score occurred by week 1 in the lurasidone 40 mg group and the olanzapine group, but not until week 3 in the lurasidone 120 mg group. In contrast, significant improvement on the CGI-S occurred by week 1 in the lurasidone 120 mg group, but not until week 2 in the lurasidone 40 mg and olanzapine groups. The reason for this discrepancy is unclear; however, the CGI-S measure reflects more than psychopathology, possibly including functional effects of cognitive improvement or a greater impact on negative symptoms.
The lurasidone 40 mg group had a lower rate of adverse events and fewer discontinuations due to adverse events than the 120 mg group. Based on number-needed-to-harm (NNH) analysis, the highest-risk adverse events (relative to placebo) in the lurasidone 40 mg group were akathisia (NNH=9.2), parkinsonism (NNH=13.3), agitation (NNH=15.2), and nausea (NNH=15.1); the highest-risk events in the lurasidone 120 mg group were somnolence (NNH=9.1), sedation (NNH=9.8), akathisia (NNH=4.5), and parkinsonism (NNH=10.8); and the highest-risk events in the olanzapine group were increased weight (NNH=6.5), sedation (NNH=8.8), dry mouth (NNH=11.1), and akathisia (NNH=15.4). These findings indicate that a higher frequency of akathisia and somnolence or sedation may be associated with the 120 mg/day dosage of lurasidone. Consistent with this, treatment with 120 mg/day of lurasidone was associated with greater treatment-emergent increases in scores on the Simpson-Angus Rating Scale and the Barnes Rating Scale for Drug-Induced Akathisia and more frequent use of medications for parkinsonism.
Our results suggest that lurasidone has a low potential for causing adverse weight and metabolic effects. There were no clinically meaningful differences between the lurasidone groups and the placebo group on levels of total cholesterol, LDL, HDL, triglycerides, insulin, glucose, or HbA
1c or on change in weight or BMI. These findings are consistent with the results from a previous placebo-controlled trial of lurasidone (
7). The pharmacological and preclinical profile of lurasidone (
20,
21) may provide a rationale for the minimal weight and metabolic effects observed in the present study. Lurasidone has no clinically relevant affinity for the receptors that have been reported to be correlated with short-term weight gain: H
1-histamine (Ki, >1000) or 5-HT
2C (Ki, 415) (
20,
22). The low rates of weight gain and metabolic disruption associated with lurasidone treatment may be particularly important given the increased prevalence of these problems in patients treated with antipsychotics (
23 –
25).
When compared with placebo, treatment with olanzapine was associated with a high proportion (34.4%) of patients with clinically meaningful weight gain as well as significant increases in levels of glucose, total cholesterol, LDL, and especially triglycerides, for which a mean increase of 50.0 mg/dl was observed. It should be noted that while all participants were required to fast per protocol, over 75% of these laboratory values were confirmed to have been obtained in a fasting state. Elevated fasting triglyceride levels are correlated with insulin resistance, since insulin-dependent lipases are typically inhibited by insulin. However, as insulin resistance worsens, increased lipolysis results in increases in free fatty acids that are hepatically transformed into triglycerides (
26). Adverse effects of olanzapine on insulin resistance and other cardiometabolic parameters have been previously documented (
24,
25). Treatment with lurasidone in this study was not associated with QTc interval prolongation >500 msec in any patient. The incidences of categorical QTc interval change ≥30 msec and ≥60 msec for patients treated with lurasidone were comparable to those for patients in the placebo group.
Treatment with lurasidone was associated with a modest dose-related increase in prolactin levels, with the 40 mg daily dose and placebo showing comparable effects, while the 120 mg daily dose resulted in greater increases than placebo at endpoint. However, the mean increase in prolactin levels on the 120 mg dose of lurasidone was less than that previously reported for risperidone, paliperidone, and conventional antipsychotics (
27).
A recent meta-analysis of atypical antipsychotics (
28) suggests that within-class differences in efficacy are relatively small, while there are large between-drug differences in safety and tolerability. The results of the present study are consistent with those findings.
Our results did not demonstrate an additional efficacy advantage for the 120 mg/day dosage of lurasidone compared with the 40 mg/day dosage. However, there was a dose-related increase in sedation/somnolence, akathisia, and use of medication for parkinsonism, as well as a modest dose-related increase in prolactin levels. The lack of an apparent dose-response relationship in this study is consistent with findings for other atypical antipsychotics (
10). The observation of comparable efficacy but an increase in some adverse effects at 120 mg is consistent with results from other clinical trials of lurasidone (
29). These findings provide support for the daily dose range of 40–80 mg for lurasidone recommended by the U.S. Food and Drug Administration. Since clinically optimal dosages of antipsychotics may change over time as medications move from investigational settings into clinical practice (
30), it remains to be seen whether wider use will alter the current clinical trial-based judgment regarding the appropriate dosage range for lurasidone.
Acknowledgments
The authors thank the participants of this study, as well as the members of the Lurasidone Study Group: in Colombia, Drs. M.M. Alzate, A. Arrieta, D. Cardona, L. Giraldo, and R. Cordoba; in India, Drs. V. Barhale, H.A. Ghandi, N.N. Raju, R. Sathianathan, Y.K. Krishanamurthy, S. Phadke, A.P. Chauhan, V. Indla, Sateesh Rao, Sathyanarayana Rao, S. Shah, H. Chandrashekhar, K. Pai, and P.S.V.N. Sharma; in Lithuania, Drs. V. Matoniene, S. Grigorjeva, V. Maciulis, and E. Mikaliunas; in the Philippines, Drs. A. Padilla, E. Reyes, E. Rondain, and J. Sy; and in the United States, Drs. G. Kaczenski, M. Lerman, R. Riesenberg, G. Brannon, R. Brenner, D. Brown, D. Feifel, J. Gilmore, S. Glass, A. Goenjian, R. Knapp, J. Kwentus, H. Meltzer, R. Mofsen, D. Nguyen, S. Potkin, S. Segal, J. Sonnenberg, T. Tran-Johnson, M. Valencerina, M. Bussel, J. Kunovac, G. Mattingly, M. Lesem, and C. Verghese.