Generalized anxiety and major depressive disorders are highly prevalent and disabling emotional disorders and are frequently comorbid with each other. Their relationship and diagnostic integrity have been the subject of considerable debate (
1–
5). On the one hand, structural analyses of comorbidity or symptomatology have pointed to anxiety and depression originating from a higher-order common “anxious/misery” factor (
6,
7), which is related to the construct of neuroticism (
8). Genetic risk for these disorders has also been shown to be similar or identical (
9,
10). We refer to this as the “common-disorder” model of anxiety and depression. On the other hand, analyses of symptomatology also support the existence of anxiety- and depression-specific phenotypes (
8,
11), and differences exist in the relationship of anxiety and depression to specific stressful life events or triggering events (
12,
13). Behavioral studies have provided evidence for automatic attentional biases for threat in anxiety and mood-congruent memory biases in depression, although such studies have rarely compared these patient groups with each other to demonstrate diagnostic specificity (
14,
15). We refer to this construct as the “independent-factor” model, in which anxiety and depression reflect separate processes. However, these findings come from complex and multidetermined measures, often lack mechanistic specificity, and are in general causally distal to the abnormalities in emotional processing that are thought to be at the core of anxiety and depressive disorders.
Neurobiological investigations of emotional processing in anxiety and depression also have yet to provide clarity regarding similarities and differences. First, almost all neuroimaging studies contrast a single patient group with healthy comparison subjects, making it difficult to compare between disorders. Second, because most neuroimaging studies either lack behavioral outcome measures or find no differences in behavior, changes in brain activation may reflect core deficits, compensation, or both. Third, anxiety and depression both involve abnormalities in core limbic regions, such as the amygdala, as well as prefrontal regulatory regions, such as the ventral anterior cingulate (
16,
17). Thus, while no canonical anxiety- or depression-related neural abnormalities have yet emerged in the literature, these design issues preclude even conclusions about commonalities between the disorders.
Understanding the nature of anxiety and depression therefore represents a paradigmatic example of the opportunities and challenges currently facing psychiatry. Such an understanding could provide insight into vulnerability and resilience factors, related in part to the heritability of these disorders; help shed light on the nosological relationships between mental illnesses; and inform treatment. Anxiety in the context of depression, for example, has been shown to predict worse response to antidepressants in general (
18), as well as in the context of specific genetic polymorphisms (
19). Yet, unless the experimental design issues mentioned above are dealt with, inferential difficulties will remain. To do so, we turned to an examination of emotional conflict processing, using a salient stimulus associated with observable and interpretable behavioral outcomes and with activation in limbic and prefrontal regions implicated in anxiety and depression (
20).
We recently described an emotional conflict task in which study subjects identify the expression of fearful or happy faces while ignoring the overlaid words “fear” or “happy” (
21–
23). In this task, emotional conflict generated reaction time interference, which was lower when an emotionally incongruent trial followed an incongruent trial than when it followed a congruent trial—an effect termed “emotional conflict adaptation” (
21,
22,
24–
27) (see Figure S1 in the data supplement that accompanies the online edition of this article). This trial-to-trial adaptation to emotional conflict reflects the operation of an emotional processing regulatory mechanism, activated by previous trial conflict, that improves performance on the current incongruent trial (
22,
28–
30) and occurs at an implicit (nonconscious) level (
23). Unlike healthy subjects, patients with generalized anxiety disorder failed to adapt to emotional conflict (
23).
Neuroimaging data in this paradigm were analyzed primarily by comparing the faster postincongruent incongruent trials to the slower postcongruent incongruent trials, drawing on the “conflict monitoring hypothesis,” which is the cognitive model that best accounts for the conflict adaptation effect (
24–
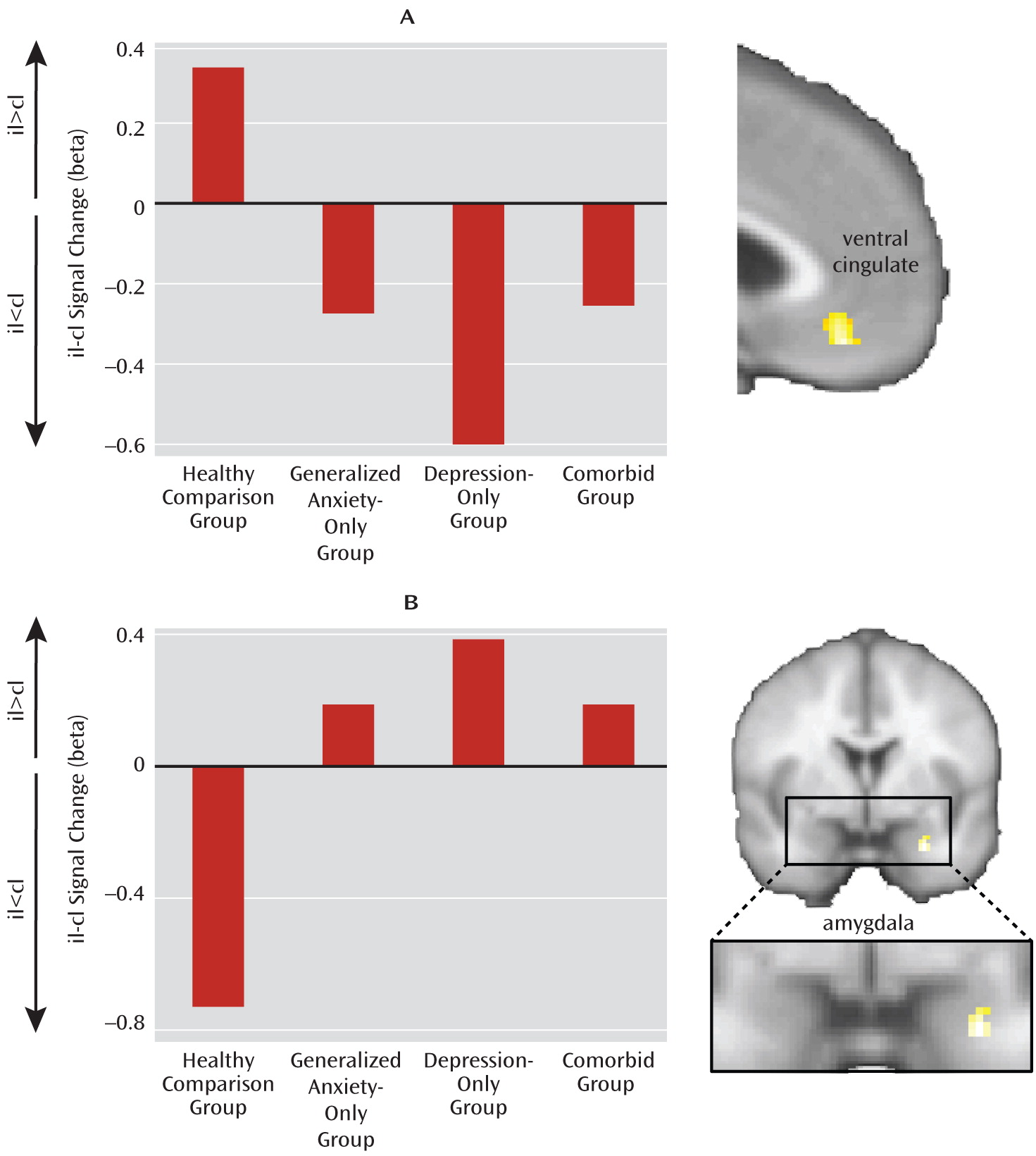
32). This model distinguishes between neural activity related to the evaluation of conflict, which is higher when conflict is greater (i.e., postcongruent incongruent trials > postincongruent incongruent trials), and dissociates it from neural activity related to the regulation of conflict. This latter function is associated with regions whose activity increases in trials when conflict is minimized through regulation (i.e., postincongruent incongruent trials > postcongruent incongruent trials). We previously examined evaluation- and regulation-related activation in both the emotional conflict task and a complementary nonemotional conflict task (gender identification of the same faces while ignoring gender label words). Regulation of emotional conflict was specifically associated with activation of the pregenual/ventral cingulate and dampening of amygdala reactivity through connectivity with the cingulate (
20–
22). By contrast, regulation of nonemotional conflict was associated with dorsolateral prefrontal activation and modulation of target-specific processing in the ventral visual cortex (
21). Notably, patients with generalized anxiety disorder failed to activate the ventral cingulate and dampen amygdala activity, consistent with their behavioral failure to adapt to emotional conflict (
23).
In this article, we extend our previous findings in generalized anxiety disorder, focusing primarily on major depression and its comparison with anxiety by reporting on emotional conflict task data from two additional groups of subjects—those with depression and no generalized anxiety disorder, and those with comorbid anxiety and depression. Specifically, we sought to determine whether results from the emotional conflict task support common-disorder or independent-factor conceptualizations of anxiety and depression, whether the results would produce evidence for behaviorally relevant dissociability of these disorders, and how differences in brain activation during implicit regulation of emotional processing in this task underlie differences in behavior.
Discussion
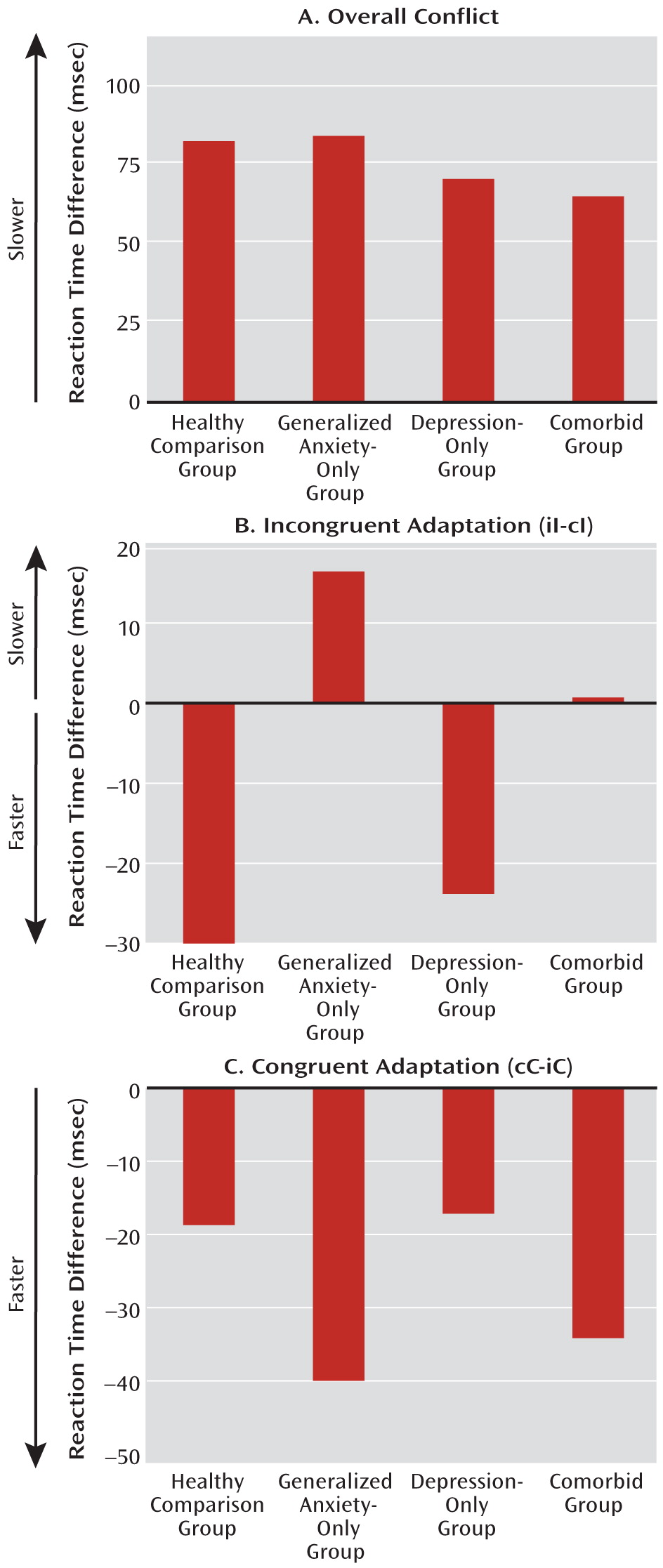
In this study, we examined implicit regulation of emotional processing in an emotional conflict task in patients with generalized anxiety disorder, major depression, or both in order to test contrasting conceptualizations of these disorders at the behavioral and neural levels. At the behavioral level, we found evidence supporting the independent-factor model, in which anxiety and depression reflect separate and dissociable processes. That is, failure to implicitly regulate emotional conflict, indexed through reaction time adaptation to emotionally incongruent trials, was perturbed in anxiety and not in depression. This effect was specific to incongruent stimuli, as adaptation to congruent stimuli was robust and intact in all groups.
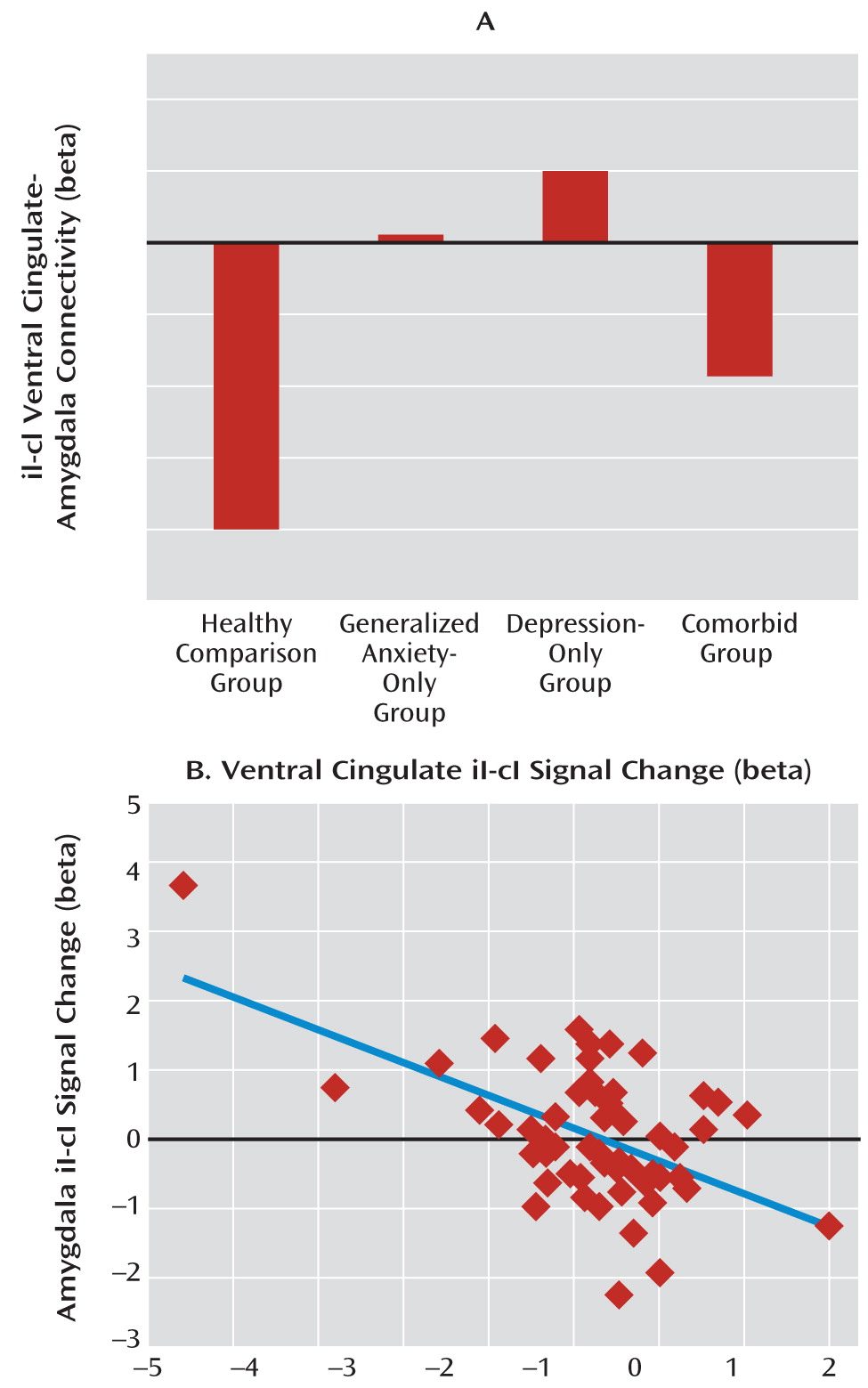
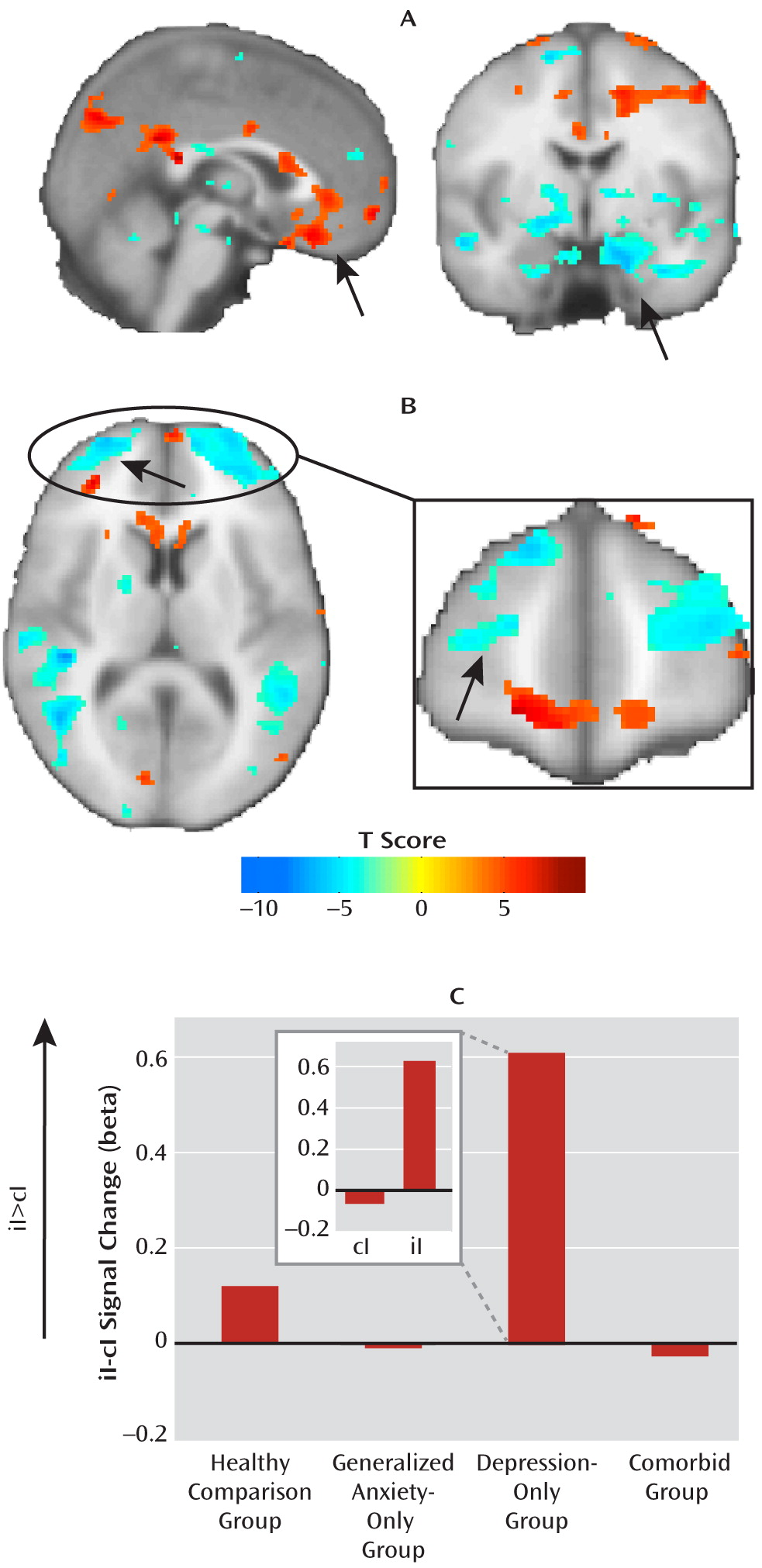
At the neural level, we found a deficit across all patient groups in the circuitry normally associated with emotional conflict adaptation—an increase in ventral cingulate activity and a dampening of amygdala activity during regulation—which supports the common-disorder model. The depression-only group, however, compensated for this deficit by activating the anterior lateral prefrontal cortex. This compensatory activation accounted for the ability of the depression-only group to regulate emotional conflict. The anxiety-specific independent-factor behavioral deficit therefore arose from a common-disorder-related abnormality in the normally used ventral cingulate-amygdala circuitry, together with an engagement of compensatory lateral prefrontal systems only in nonanxious depression. The specificity of this compensation for nonanxious depression is striking and suggests that it may be related to aspects of depression not shared with anxiety (e.g., anhedonia). Nonetheless, since overall depression levels were the same for the depression-only and comorbid groups, the mere presence of features of depression unrelated to anxiety was insufficient to result in compensation when anxiety was also present, pointing to a conditional relationship between features of depression associated with compensation and anxiety.
To date, few functional neuroimaging studies have compared emotional processing in anxiety and depression, and none have done so in adults. One recent study (
51), focusing largely on amygdala activation in adolescents in response to emotionally expressive faces under a variety of attentional conditions, found evidence for both the common-disorder and independent-factor models, which were further moderated by the specific attentional demands of the task. A pediatric study (
52), using only one attentional condition during fearful face viewing, found divergent amygdala responses in anxiety and depression. As our data illustrate, parallel investigations of multiple diagnostic groups provide essential and novel information regarding the similarities and differences between disorders, which is not possible through extrapolation from published studies on single-disorder cohorts, and which may allow a reconceptualization of mental illness along dimensions determined by neural and behavioral systems rather than complex symptom expression (
53).
Our data indicate that even seemingly simple behavioral measures are themselves determined by several simultaneously operating neural processes, suggesting ultimately that an understanding of anxiety and depression must consider these multiple “layers” of abnormalities that together drive a more complex outcome measure, such as symptoms or diagnosis. One of these neural layers is the shared deficit in ventral cingulate-amygdala activation and functional connectivity. In light of the findings that anxiety and depression share similar genetic risk factors (
9,
10), which may be an important aspect of their similarity (
5), it is noteworthy that several lines of genetic risk investigations also converge on ventral cingulate-amygdala changes. Polymorphisms in the serotonin transporter gene, for example, have been associated with an elevated risk for depression through an interaction with negative life events (
54), are related to the general negative affectivity factor of neuroticism (
55), and result in differences in amygdala and cingulate activation, structure, and connectivity (
56–
59). Similar findings have been reported for polymorphisms in other genes involved in serotonin biosynthesis or signaling, as well as other monoaminergic genes, such as the monoamine oxidase A gene, as well as neurotrophins, such as brain-derived neurotrophic factor (
59–
62). Individuals who are at risk for depression by virtue of a family history of depression also show abnormalities in amygdala and cingulate activation (
63,
64). Finally, a recent structural imaging study (
65) of patients with depression, a mixture of anxiety disorders, or comorbid depression and anxiety disorders found common-disorder volumetric reductions across all groups in an anterior cingulate region near our ventral cingulate cluster, although the functional consequences of these abnormalities were unknown because of the nature of structural imaging.
Despite this common ventral cingulate-amygdala deficit, the depression-only group was able to compensate, activating the anterior lateral prefrontal cortices and adapting to emotional conflict. A recent neuroimaging meta-analysis (
66) found that these prefrontal regions were very likely to coactivate with a canonical fronto-parietal executive control network across a wide range of cognitive tasks. Aberrant engagement of this cognitive control system suggests that depression-only participants were somehow able to adapt by using a different neural strategy. Indeed, the surprising relationship between ventral cingulate-amygdala activation and adaptation in the depression-only group (better adaptation associated with more neural dysfunction) suggests that compensatory circuitry was engaged at the expense of the normal pathway implicated in adaptation, possibly because the two may normally be in competition. This is striking for several reasons. First, it suggests that intact performance in this task can be achieved despite abnormalities in the core circuitry implicated in conflict adaptation and thus that improvements in performance (e.g., as a consequence of treatment) need not necessarily operate by reversing the baseline deficit. Second, although not formally addressed in these patients, we have reported (
23) that participants are unaware of the conflict adaptation effect, indicating that it is an implicit process. Thus, the ability of depressed patients to adapt likely reflects operation of an implicit compensatory process (lateral prefrontal activation). These findings also deepen our understanding of implicit emotion regulation and further emphasize the importance of understanding these processes for investigating psychopathology. In parallel with the present study, we also recently reported compensatory coupling of lateral prefrontal regions with the amygdala in patients with generalized anxiety disorder using resting-state imaging (
67). This compensation did not appear to affect that group's performance in this task, and thus it may have functional consequences only when participants process or regulate emotion explicitly, akin perhaps to the attention-demanding process of worry.
Together, our findings bring to greater relief the idea that most, if not all, psychiatric disorders, having their origins years before symptom expression, likely reflect a complex interplay between core deficits and compensatory systems. A more comprehensive understanding of anxiety and depression in that light will require testing the same subjects in multiple tasks that systematically probe implicit and explicit emotional processes, include longitudinal designs or samples across the lifespan whenever possible, and examine the influences of treatment, in order to determine what the consequences of core deficits are and to identify the functional consequences of compensation. Doing so may in turn inform our understanding of the etiology of these disorders and the routes for improving their treatment.
Finally, alternative conceptualizations of anxiety and depressive psychopathology have called for use of a dimensional, instead of a categorical, diagnostic approach (
40,
41). As noted, all of our key category-based behavioral findings were independent of individual differences in symptom scale scores, which suggests that these commonly used questionnaires do not adequately capture the behavioral dissociation of anxiety and depression in our task, despite the fact that symptom measures can discriminate between groups (
Table 1). Moreover, we suggest that dimensional measures of complex symptoms may provide only limited insight into the nature of psychopathology given the complex layering of deficits and compensations at the neural level, as illustrated in our data.
Several limitations of this study are also important to note. First, this was a cross-sectional study, and thus it is impossible to determine whether symptom changes over time may be associated with dynamic changes, for example, in anterior prefrontal activation. This might mean that loss of compensation may result in comorbidity or that enhancements in compensation may lead to lessening of anxiety in the context of depression. Further longitudinal imaging will be required to test this hypothesis. Second, we did not include in this study patients taking medications. While this likely means that we excluded the more severe cases, inclusion of medicated patients would significantly confound brain activation results, as medications for anxiety and depression alter brain activation in precisely the regions under study. Finally, it might be argued that the intact adaptation in the depression-only group is related to these individuals being less ill or the stimuli being more anxiety- than depression-relevant. However, we find these alternative explanations to be unlikely. The depression-only group was more depressed (but less anxious) than the anxiety-only group and had similar scores on the State-Trait Anxiety Inventory, the Beck Depression Inventory, and the anhedonic depression subscale of the Mood and Anxiety Symptom Questionnaire as the comorbid group (
Table 1). Moreover, they were able to adapt not by virtue of being able to engage ventral cingulate-amygdala circuitry, but despite a profound deficit in this circuitry. In summary, our results provide an exciting framework for future work investigating implicit emotion regulation in psychopathology and the nature of anxiety and depression.