Schizophrenia is a severe and common neuropsychiatric disorder with an estimated lifetime prevalence approaching 1% worldwide (
1). The essential characteristics of this disorder include various psychotic symptoms such as delusions and hallucinations, affective response, social withdrawal, apathy, and cognitive impairment (
2). Family, twin, and adoption studies have unequivocally shown a strong genetic component in schizophrenia, with heritability estimates of about 80% (
3). Despite such high heritability of schizophrenia, the underlying genetic risk factors have yet to be identified. Numerous genetic association studies of schizophrenia have been reported, and numerous candidate genes have been proposed through linkage analyses, candidate gene association studies, and genome-wide association studies (GWAS) (
4–
7). Many of them, however, await satisfactory replications in different populations and functional verification of the relevant genetic variants.
ZNF804A, a novel schizophrenia susceptibility gene on chromosome 2q32.1, was first identified by GWAS in U.K. populations with large-scale follow-up replications (
8). The risk single-nucleotide polymorphism (SNP), rs1344706, achieved genome-wide significance, and it was further confirmed by subsequent independent replication studies as well as meta-analyses in major populations (
9–
14). Through fine mapping analysis, Williams et al. (
10) observed multiple common SNPs within ZNF804A that were strongly associated with schizophrenia. Meanwhile, Steinberg et al. (
9) searched for large copy number variations in 5,408 psychiatric patients (including those with schizophrenia, bipolar disorder, anxiety, and depression) and 39,481 healthy comparison subjects and identified three copy number variations covering part of ZNF804A in psychiatric patients but not in comparison subjects (p=0.0016). Thus, ZNF804A is a promising risk gene for schizophrenia, especially in European populations.
Studies of Chinese populations, however, have been inconsistent. O'Donovan et al. (
8) and Steinberg et al. (
9) failed to replicate the association of rs1344706 with schizophrenia in Han Chinese (p=0.166 and p=0.62, respectively), but Zhang et al. (
11) did replicate the association (p=0.00083). This suggests that either ZNF804A may not be a risk gene for schizophrenia in the Chinese population, or there are other risk SNPs. Additionally, it was reported that when considering disease status, the expression of ZNF804A was higher in patients than it was in comparison subjects but was not significantly different (p=0.107). When considering rs1344706 allele status in comparison samples only, expression was significantly higher from the associated allele (p=0.033) (
13). We speculate that those SNPs located in the promoter region of ZNF804A may affect ZNF804A expression and eventually contribute to the pathogenesis of schizophrenia.
To test whether ZNF804A is a risk gene for schizophrenia in the Chinese population, we first examined rs1344706 in two independent case-control samples collected from southwestern China. We then tested five ZNF804A promoter SNPs (rs359895, rs10497655, rs13026173, rs11888068, and rs1021042) in these samples, in which we observed two SNPS (rs1021042 and rs359895) that were significantly associated with schizophrenia. Finally, we investigated the functional impact of rs359895 on transcription factor binding affinity and promoter activity.
Method
Participants
We recruited two case-control samples independently from the southwestern Chinese cities of Yuxi (502 patients with a mean age of 38.5 years [SD=10.4]; 694 comparison subjects with a mean age of 37.1 years [SD=6.8]) and Kunming (404 patients with a mean age of 36.3 years [SD=8.7]; 607 comparison subjects with a mean age of 36.6 years [SD=7.0]). The patients were diagnosed with schizophrenia according to ICD-10 (Yuxi) or DSM-IV (Kunming) criteria. We collected detailed information on the course of the clinical disorder, age at onset, symptoms, and family history of psychiatric illnesses. Potential participants with a history of alcoholism, epilepsy, neurological disorders, or drug abuse were excluded from the study. Meanwhile, unrelated healthy volunteers were recruited from the local communities as comparison subjects. These individuals were asked to provide detailed information about their medical and family psychiatric histories. Those who had a history of psychiatric disorders, psychiatric treatment, alcohol dependence, or drug abuse or a family history of psychiatric disorders were excluded.
The patients and comparison subjects were of Han Chinese origin from the Yunnan province of southwestern China, and they were all unrelated. All participants provided written informed consent, and the research protocol was approved by the internal review board of the Kunming Institute of Zoology at the Chinese Academy of Sciences.
SNP Selection
SNP selection was based on previous studies and our HapMap data analysis (
8–
13). For the initial screening in the Yuxi sample, a total of six SNPs were selected. We first selected rs1344706, which has been identified by GWAS in European populations (
8). We then examined the linkage disequilibrium pattern of the ZNF804A promoter region (about 10 kb) in the Chinese population using data from HapMap. We initially selected five tagging SNPs spanning the promoter region of ZNF804A (rs1021042, rs13026173, rs359895, rs10497655, and rs359894) with the use of the tagger procedure implemented in Haploview (
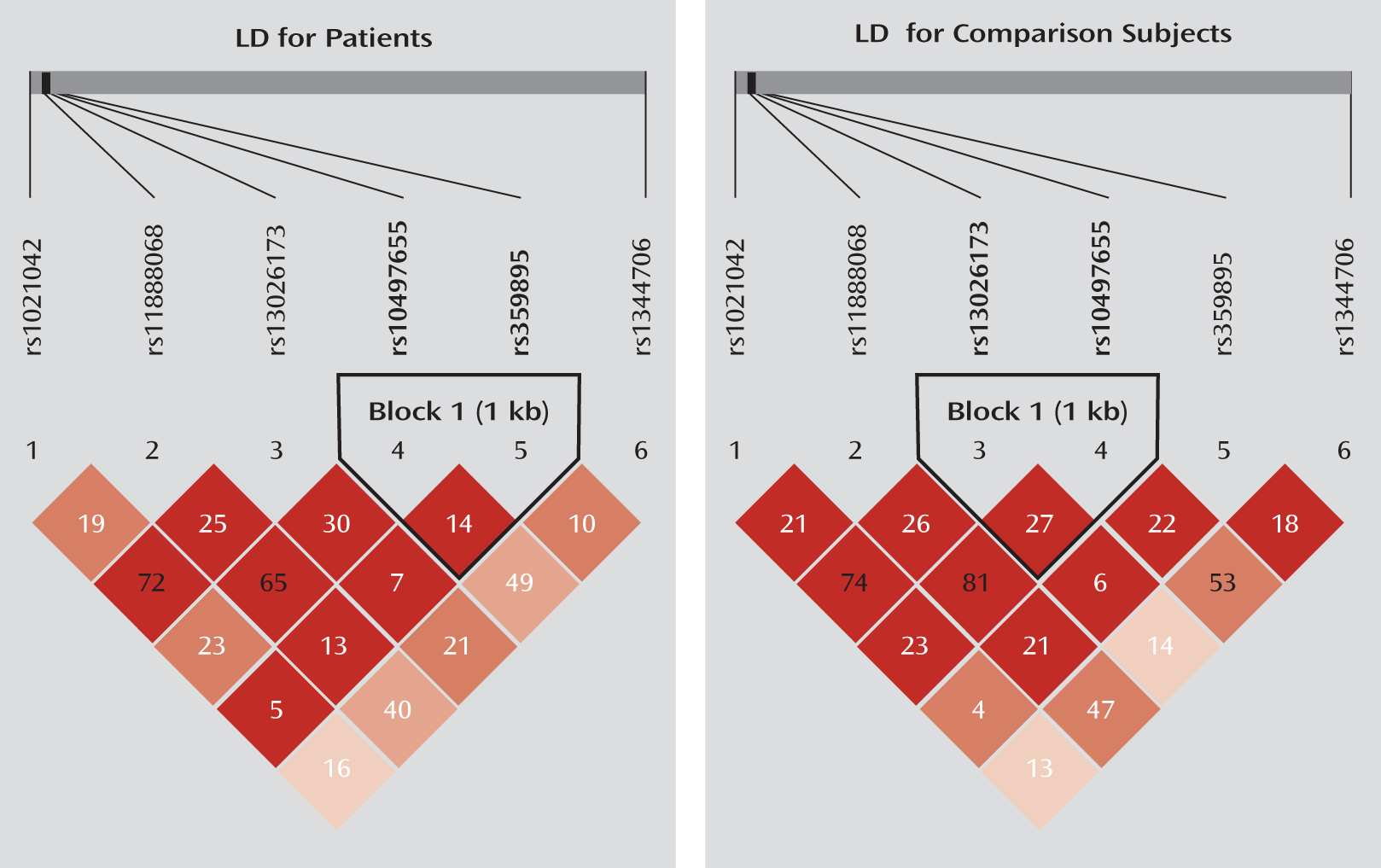
15). Since the minor allele frequency of rs359894 is less than 0.05 in Han Chinese (minor allele frequency=0.022, shown in HapMap), we excluded this SNP from further analysis and selected another one (rs11888068) close to rs359894. All the selected SNPs are biallelic with the minor allele frequency greater than 5%. In total, six SNPs were selected for screening the Yuxi sample (see Figure S1 in the data supplement that accompanies the online edition of this article). For the replication analysis in the Kunming sample, three SNPs were tested, including rs1344706 and the two SNPs (rs359895 and rs1021042) that showed significant associations with schizophrenia in the Yuxi sample. The linkage disequilibrium map of the tested SNPs in the Yuxi sample is shown in
Figure 1, and the linkage disequilibrium maps of the ZNF804A promoter SNPs downloaded from the HapMap database are shown in Figure S1 and Figure S2 in the online data supplement. The SNP information is shown in
Table 1.
SNP Genotyping
Venous blood was collected from all participants, and genomic DNA was extracted from the blood sample using the standard phenol-chloroform method. DNA samples of the patients and comparison subjects were randomly distributed in the DNA sample plates. All the selected SNPs were genotyped by the SNaPShot method as described in our previous study (
16). Details of all primers and assay conditions are available on request. The SNP genotype calls were automatically performed using GeneMapper 4.0 (Applied Biosystems) and verified manually. To make sure of the accuracy of genotyping, we conducted bidirectional sequencing of 100 randomly selected individuals, and no genotyping errors were observed. The genotyping success rate for the six tested SNPs was 98.4%.
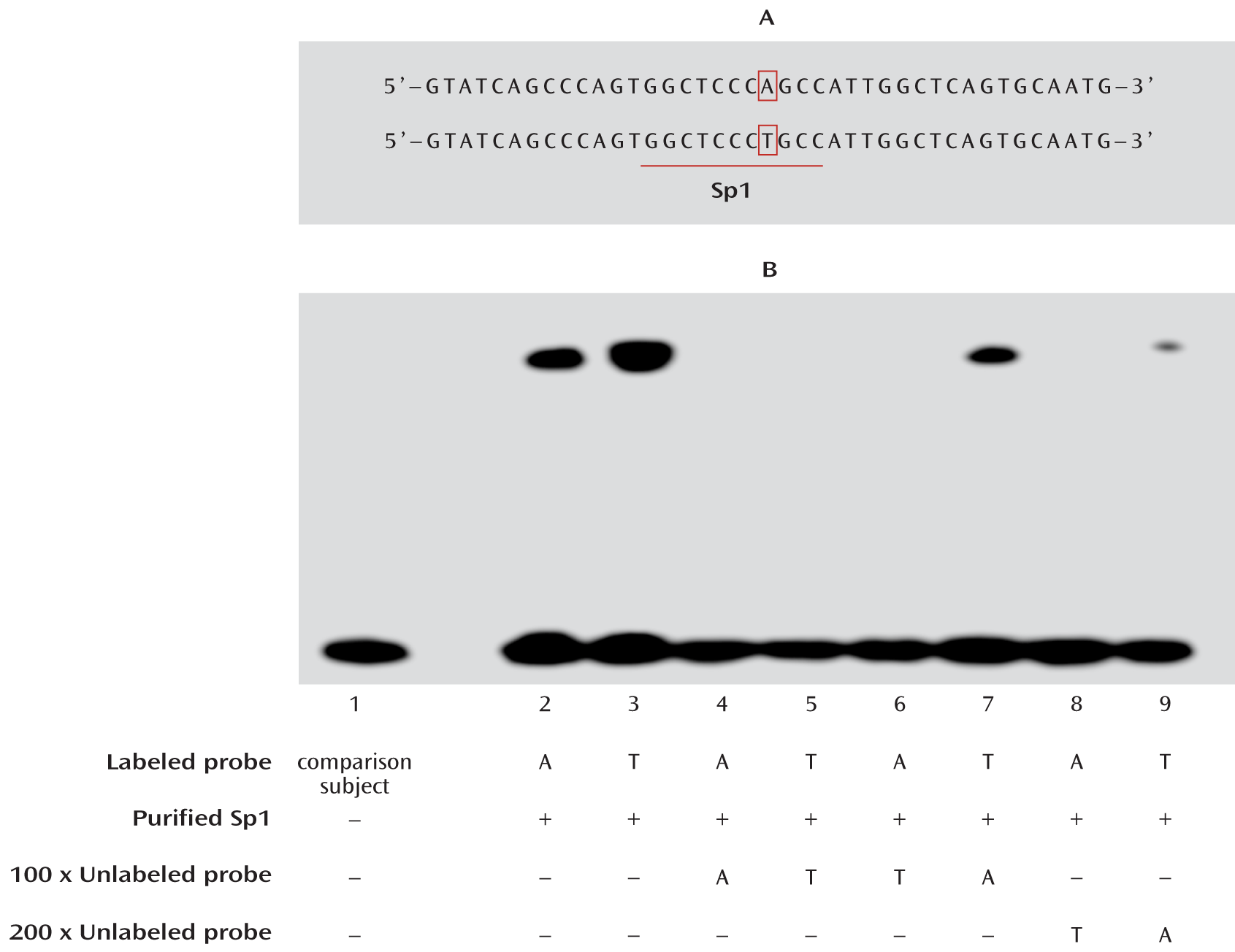
Electrophoretic Mobility Shift Assay
For functional prediction of the candidate SNPs, we used the online software program AliBaba (
www.gene-regulation.com/pub/programs.html#alibaba2) to predict the DNA-binding motifs in the ZNF804A promoter region. A 100% match to the matrix yields a maximum score of 1.00, and a matrix similarity score greater than 0.80 is considered a good match.
Electrophoretic mobility shift assay was performed using the gel shift assay system (Pierce Protein Research, Rockford, Ill.) under the guidelines provided. The single-strand oligonucleotides were 3′-end biotin labeled and then annealed to form double strands. The binding reaction contained purified recombinant Sp1 protein (Alexis Biochemicals, San Diego), 10× binding buffer, and, if needed, unlabeled competitors; the labeled probes were added after 20 minutes, and samples were incubated at room temperature for a total of 30 minutes. After incubation, samples were separated on a native 6% polyacrylamide gel and then transferred to a nylon membrane. The positions of biotin end-labeled oligonucleotides were detected by a chemiluminescent reaction with streptavidin-horseradish peroxidase. The nucleotide sequences of the double-stranded oligonucleotides with either A or T allele were:
A allele: 5′-GTATCAGCCCAGTGGCTCCCAGCCATTGGCTCAGTGCAATG-3′
T allele: 5′-GTATCAGCCCAGTGGCTCCCTGCCATTGGCTCAGTGCAATG-3′
Luciferase Reporter Assay
To construct ZNF804A promoter, we amplified fragments encompassing nucleotides from –1089 base pairs (bp) to +65 bp (relative to transcription start site +1) from two individual homozygotes with respect to the corresponding genotypes (TT and AA) for rs359895. Then the amplified fragments were cloned into the pGL3-basic plasmid vectors. We verified all recombinant clones by bidirectional DNA sequencing to make sure no de novo mutation was introduced. These plasmids were all accurately quantified with an Eppendorf (Hamburg) BioPhotometer, and equal amounts of the plasmids were used for transfection. The reporters containing either T allele or A allele were transiently cotransfected into HEK293T and HeLa cells together with pRL-TK plasmid (a standard reporter). After a 36-hour incubation, we collected the cells and measured luciferase activity using the Dual-Luciferase Reporter Assay System (Promega Corporation, Madison, Wisc.). All assays were performed in at least three independent experiments with a minimum of five replications.
Statistical Analysis
We tested for Hardy-Weinberg equilibrium using Haploview 4.1 by examining the genotypic distributions of ZNF804A SNPs in each sample. All of the SNPs genotyped in this study were in Hardy-Weinberg equilibrium. Linkage disequilibrium between paired SNPs was estimated by Haploview using the r
2 algorithm. Allelic and genotypic associations were accessed with PLINK (
17). Since the SNPs may not be totally independent because of linkage disequilibrium, to avoid type II error when applying the Bonferroni correction, we corrected the p values with a max(T) permutation procedure implemented in PLINK, using the “–mperm” option (N=1,000,000), which takes a single parameter (the number of permutations to be performed). This is achieved by comparing each observed test statistic against the maximum of all permuted statistics (i.e., over all SNPs) for each single replicate. We calculated significance for the combined samples (the Yuxi and Kunming samples) using the Cochran-Mantel-Haenszel test and conditioning by site as implemented in PLINK. The 95% confidence intervals (CIs) of odds ratios were calculated with an online tool (
http://faculty.vassar.edu/lowry/odds2x2.html). For meta-analysis of rs1344706 in the five Han Chinese samples independently collected from Shanghai, Xi'an, Sichuan, Yuxi, and Kunming (from O'Donovan et al. [
8], Steinberg et al. [
9], Zhang et al. [
11], and the present study), because there is genetic heterogeneity caused by the data from Zhang et al. (
11), we used the Mantel-Haenszel method with the random-effects model. For the meta-analysis excluding the sample from Zhang et al. (
11), we used the Mantel-Haenszel method with the fixed-effects model, and the analysis was conducted in RevMan, version 4.2 (
http://ims.cochrane.org/revman/download/revman-4). Haplotype frequencies and association analysis were estimated using the HaploStats Package in the R environment (
18). Correction for multiple haplotypes and global analysis were performed with 20,000 permutations (
18). The power analysis was performed using the G*power software program (
19). To avoid the false positive association caused by potential population stratification, we used the software program Structure to perform population stratification analysis in our samples (
20,
21). For the reporter gene assay data analysis, we used two-tailed t tests to conduct statistical assessment.
Discussion
In 2008, O'Donovan et al. (
8) undertook a GWAS of 479 U.K. schizophrenia patients and 2,937 comparison subjects, with follow-up replications in approximately 17,000 subjects, and identified rs1344706 within ZNF804A as significantly associated with schizophrenia. Subsequently, the association of rs1344706 with schizophrenia was consistently reported by the International Schizophrenia Consortium, the Irish Case-Control Study of Schizophrenia, and the SGENE-plus Consortium (
9,
13,
23). Further investigations have indicated that there are allelic differences of rs1344706 in ZNF804A expression and nuclear protein binding affinity (
10,
13,
24). Since it is located in the intron region, however, there is still the possibility that rs1344706 is a tagging SNP linked to the causative variant at this locus. To address this, Williams et al. (
10) sought to localize the association signal at this locus through a process of genomic resequencing and fine-scale mapping. After detailed association analysis, rs1344706 remained one of the most strongly associated markers in ZNF804A. In addition, a meta-analysis of rs1344706 in 18,945 schizophrenia patients and 38,675 comparison subjects (
10) again supported the association between rs1344706 and schizophrenia (p=2.5×10
–11). These data have strongly indicated that rs1344706 is a promising risk SNP for schizophrenia, especially in European populations.
In contrast, the association of rs1344706 with schizophrenia in Han Chinese is inconsistent among studies. No significant association between rs1344706 and schizophrenia in Han Chinese was observed in the replication studies by O'Donovan et al. (
8) and Steinberg et al. (
9), while Zhang et al. (
11) reported significant associations (p<0.001). In the two independent samples tested in our study, we failed to detect significance for rs1344706, and this finding was further confirmed by the meta-analyses of Chinese populations (p=0.38). These analyses suggest that rs1344706 is probably not a risk SNP for schizophrenia in Han Chinese.
Notably, the T allele frequency of rs1344706 in our comparison subjects was 0.509, which is similar to the frequencies reported by O'Donovan et al. (frequency=0.514) and Steinberg et al. (frequency=0.546) as well as the frequency in Han Chinese from the HapMap database (frequency=0.524). It is quite different, however, from the frequency reported by Zhang et al. (frequency=0.457), implying that there is either sampling bias in Zhang et al. (
11) or potential genetic heterogeneity among regional Han Chinese populations as reflected in the results of heterogeneity tests in the meta-analysis. Moreover, the inconsistent association of rs1344706 with schizophrenia between Chinese and European populations also suggests genetic heterogeneity of ZNF804A sequence variations among continental populations. The population-specific factors (population history, differences in genetic structure, environmental exposure, diet, and culture) may play an important role in the observed inconsistency of replications.
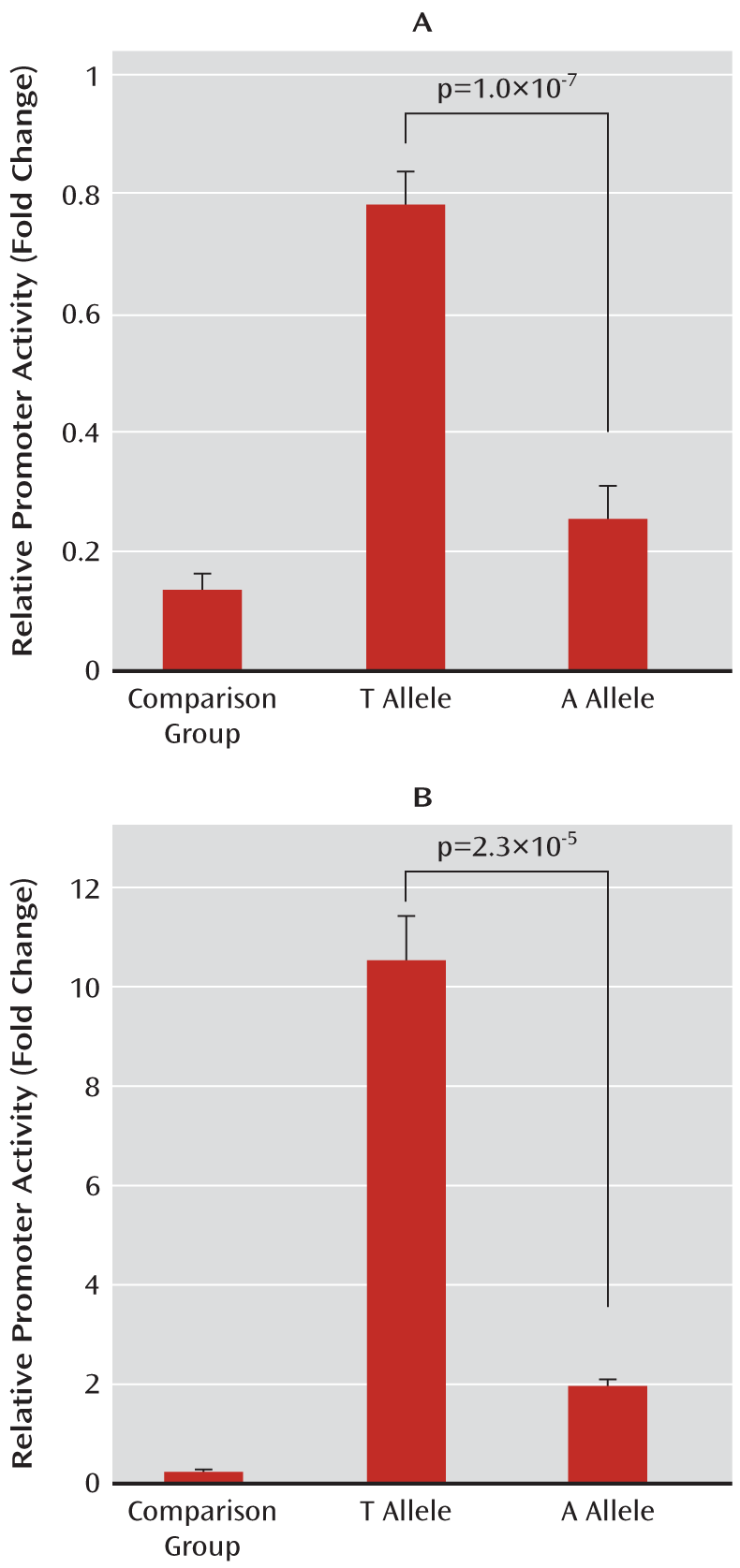
We identified a novel functional SNP (rs359895, minor allele frequency=0.2198) that is strongly associated with schizophrenia in two independent Han Chinese samples (p=1.0×10
–5), and the haplotype analysis further supported these associations (global p<1.0×10
–8). Our functional assays indicated that rs359895 can influence the affinity of the Sp1 binding sequence and that the T allele has a higher binding affinity to Sp1, leading to a higher promoter activity and higher ZNF804A expression than the A allele. This is consistent with previous findings in which the risk allele of rs1344706 showed significantly higher ZNF804A expression in comparison subjects (p=0.033), and an elevated expression was detected in the prefrontal cortex of schizophrenia patients relative to comparison subjects, although it did not reach significance (
13). Additionally, another newly identified SNP (rs1021042) was significantly associated with schizophrenia in our samples (p=3.0×10
–6), but it showed no potential function based on the prediction. The SNP rs1021042 is in low linkage disequilibrium with rs359895 in both the patients and comparison subjects as well as in the HapMap Han Chinese in Beijing (CHB) (r
2≤0.05), indicating that the association of rs1021042 with schizophrenia is likely independent.
The previous fine-mapping study of ZNF804A by Williams et al. (
10) reported many SNPs that are strongly associated with schizophrenia; however, rs359895 and rs1021042 were not tested. Interestingly, according to the HapMap data, there is a large heterogeneity for rs359895 among continental populations. The T (risk) allele of rs359895 is prevalent in East Asian populations (frequency=0.800 in Chinese and frequency=0.920 in Japanese), but much less frequent in Europeans (frequency=0.408) and Africans (frequency=0.217), and the linkage disequilibrium structures of the ZNF804A promoter region also differ among major populations (see Figure S2 in the online data supplement). Therefore, whether rs359895 is also associated with schizophrenia in European populations has yet to be tested.
Despite many genetic association studies, the function of ZNF804A is still unknown. ZNF804A, consisting of four exons and encoding a protein of 1,210 amino acids, is expressed in the brain (
25). The amino acid sequence contains a C2H2-type domain characteristic of the classical zinc-finger (ZnF) family of proteins, which may interact with other types of molecules and have many roles in cellular function (
25). Interestingly, the mouse homologue of ZNF804A, zfp804a, has recently been reported as a target for HOXC8, suggesting that ZNF804A may be involved in the regulation of early neurodevelopment (
26). Additionally, recent investigations have indicated ZNF804A may also act on neural activation and cognitive performance (
12,
27–
29).
The data we present here are limited, and we are cautious in the interpretation of our results. First, we tested only the ZNF804A promoter SNPs, and there are other ZNF804A risk SNPs in Chinese populations that have yet to be studied, although the importance of promoter regions in identifying risk genetic variants for schizophrenia has been verified (
30,
31). Second, although we tested two independent samples (more than 2,000 individuals) in this study, our sample size is still relatively small compared with the current large-scale genetic studies (
8–
10,
32), and the association of rs359895 needs to be tested in more samples.