Heredity is intimately related to the history of psychiatry. Clinical observations by early physicians noted the tendency of mental illnesses to run in families. In the 20th century, these anecdotes were systematically evaluated, and some were confirmed in rigorous twin, family, and adoption genetic epidemiological studies. This exceptional body of evidence provided a major etiological clue for the field: common psychiatric disorders have a moderate to strong tendency to run in families largely due to genetic inheritance (
1,
2).
For instance, in 1946 Kallmann published an influential twin study of schizophrenia in this journal (
3). Kallmann was a psychiatrist and the fourth president of the American Society for Human Genetics. Kallmann’s study of 691 twin pairs was the largest in the field for nearly four decades. Reanalysis of these data (
4) yielded an estimate of the heritability of schizophrenia (91%) that was higher than estimates from more recent national-scale studies (60%−65%) (
5,
6). Although Kallmann’s speculation that schizophrenia was due to an autosomal recessive mutation has been disproven, the concluding line of his article remains exceptionally important, stating that a genetic theory of schizophrenia is “equally compatible with the psychiatric concept that schizophrenia can be prevented as well as cured.”
We now know that these genetic effects are relatively small and nondeterministic: most people with a strong family history are not themselves affected (as is also observed for most complex biomedical diseases). Moreover, most psychiatric disorders do not “breed true.” For example, the immediate relatives of people with schizophrenia have increased risks not only for schizophrenia but also for multiple other conditions (e.g., bipolar disorder, major depressive disorder, and autism). The diverse clinical manifestations and variable course observed for many common psychiatric disorders are consistent with complex and relatively small genetic effects. For adult-onset common psychiatric disorders in particular, development is often within normal limits, although there is often some impairment of higher components of cognition.
In the last decade it has become technically and economically feasible to interrogate the genome directly with increasing resolution and completeness. Instead of indirectly studying the heredity of psychiatric disorders (e.g., through studies of pedigrees, twins, or adoptees), we can now evaluate the genomes of case and control subjects at several levels of precision quickly and inexpensively. Indeed, heritability itself can be assessed directly from genome-wide genetic data (
7,
8).
By carefully evaluating the successes and failures of psychiatric genetics in the past three decades, we now have a solid fix on how to dissect the “family history risk factor” into far more precise and mechanistic components. We can identify genetic variants that contribute to risk and are moving toward understanding the mechanisms by which they act. The field has learned an enormous amount and is poised to make fundamental advances that could profoundly improve understanding.
This review provides an update on what we have learned and puts forth an agenda for the next 5 years. A key lesson was the need for a global community effort in psychiatric genetics because the required sample sizes are far beyond the reach of any single group. To enable these studies, in 2007 we formed the Psychiatric Genomics Consortium (PGC) (
http://www.med.unc.edu/pgc). Our overarching goal is to deliver actionable knowledge, i.e., genetic findings whose biological implications can be used to improve diagnosis, develop rational therapeutics, and craft mechanistic approaches to primary prevention.
Getting up to Speed in Genetics
In 2009, the PGC published three foundational articles regarding genome-wide association studies (GWAS) (
9–
11). GWAS is a genomic study design that focuses on the impact of common genetic variation in almost all genes in the human genome. The initial PGC articles covered the core concepts, history, rationale, genomic assays, statistical analysis, interpretative framework, and importance of cross-disorder studies in psychiatry. Full background of the terminology, core concepts, and strategy of GWAS can be found in these articles. Basic terms are defined in Table S1 in the
data supplement accompanying the online version of this article, and a “primer” has been published previously (
12).
Clarity in Retrospect
A key unknown was genetic architecture, particularly the sizes of the underlying genetic effects. A decade ago these were unknown and subject to considerable speculation, with hypotheses suggesting that genetic discovery for psychiatric disorders would be anywhere from highly tractable to impossible. If the genetic effects were few, common, and large, relatively modest sample sizes would be sufficient. A few early studies hinted that small samples might suffice (e.g., studies of the effects of
APOE on Alzheimer’s disease [
13] or
CFH on age-related macular degeneration [
14]), and these may have led to expectations that gene discovery would be straightforward.
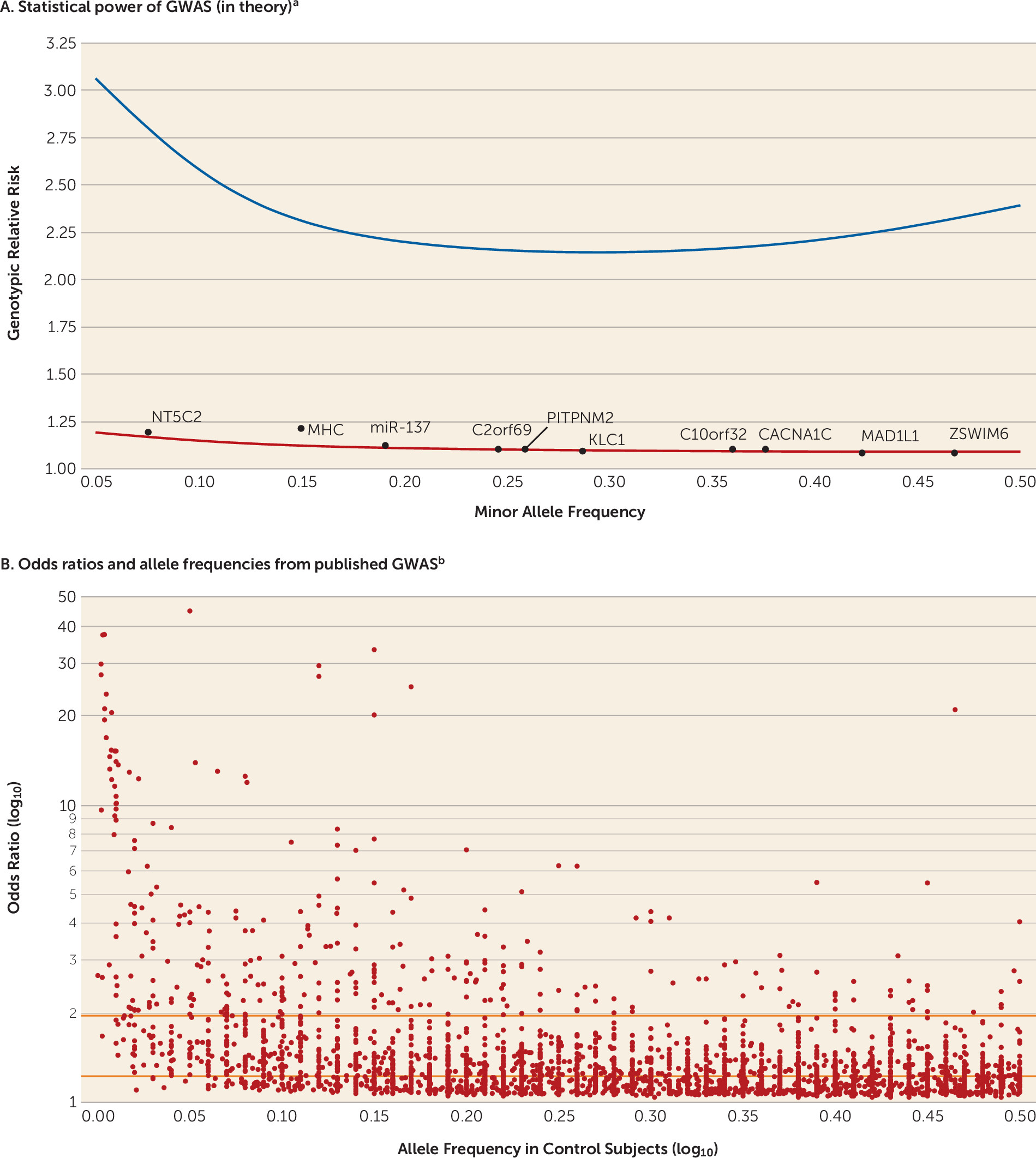
The power calculations are not difficult: for a given number of case and control subjects (plus assumptions of allele frequency, genetic model, significance threshold, and power), it is easy to compute the minimum detectable genotypic relative risk. For example,
Figure 1A shows the 90% power curve for a GWAS of 1,000 cases and 1,000 controls.
Like most investigators in human complex disease genomics, we had limited data to allow us to narrow bounds on the search space. We quickly learned that optimistic assumptions of large genetic effect sizes for these disorders were incorrect. The initial GWAS for psychiatric disorders had sample sizes of approximately 1,000 cases, enabling excellent power to detect a genotypic relative risk ≥2.5. However, these effects were not found for schizophrenia (
16), bipolar disorder (
17), major depressive disorder (
18), or attention deficit hyperactivity disorder (ADHD) (
19).
Figure 1A also shows the 90% power curve for the most successful GWAS of any psychiatric disorder (37,000 schizophrenia cases) (
15), and only two of 128 independent loci had genotypic relative risks ≥1.2. Compellingly, we can now demonstrate that common genetic variants with genotypic relative risks above ∼1.24 for schizophrenia can be excluded with about 100% power.
Genetic effects that are common and large are unusual for human diseases and traits studied by means of GWAS (
Figure 1B). They are occasionally found for less complex conditions that can be assessed with exceptional precision (e.g., infectious diseases, rare adverse drug reactions, and eye diseases). To our knowledge, the largest common genetic variant associations observed to date in psychiatry are for alcoholism in people of East Asian ancestry (genotypic relative risk, ∼6.2) (
20) and clozapine-induced agranulocytosis (genotypic relative risk, ∼5.3) (
21).
Genetic Architecture and Models of Disease
Elucidation of the genetic architecture underlying these disorders is the major goal of the PGC. How many susceptibility or protective variants are there? What are their frequencies and effect sizes? How do they exert their effects? Do these variants interact with one another or with environmental risk factors? Crucially for biological understanding, which genes are affected by these variants?
It is heuristically useful to consider the “bookends.” The extreme models are that psychiatric disorders are caused by 1) the cumulative impact of hundreds or thousands of common genetic variants each of small effect (common disease/common variant model) or 2) many different gene-disrupting variants of strong effect (multiple rare variant model). In the latter model, every person with a serious psychiatric disorder would have a strong effect variant, and these would cluster in a set of genes important to brain development and function.
These models were passionately debated. Some authors expressed profound hope that the multiple rare variant model was broadly explanatory (
22–
24). Others favored a common disease/common variant model, arguing that psychiatric phenotypes are comparatively subtle. Most investigators were agnostic. The PGC wished to design studies that would be informative whatever the underlying model (
9).
Initial Strategy
A consistent lesson from the history of psychiatric genomics was that these are very hard problems: any search is going to be far more difficult than anticipated. Although we were hopeful that the initial GWAS might deliver insights, we created the PGC in order to hedge our bets; we needed a framework to aggregate data across studies with exceptional care and rigor if we were to progress. A critical step was to convince all groups that sharing individual data was essential—this is a foundational principle of the PGC and allows optimal quality control and analysis.
Moreover, to ensure progress, an “open science” perspective was required. Genome-wide summary statistics of all PGC analyses are available for widespread use (
http://www.med.unc.edu/pgc), and the vast majority of PGC genotype data that can be deposited are available to qualified researchers in a controlled-access repository. We recently have made available a list of the top loci from PGC analyses (both published and in preparation).
These early strategic decisions proved important, as results from the first wave of psychiatric GWAS, circa 2008, were unimpressive. Although we were careful not to hype GWAS (
9,
10), some prominent commentators voiced strong doubts about its value—even though careful review of the early results showed unequivocal indications of genetic effects. The first-wave studies were simply underpowered, and combining studies to increase power was logical. Nevertheless, we persisted, and a 2012 letter signed by 96 psychiatric genetics investigators (“Don’t give up on GWAS”) anticipated the utility of GWAS should sample sizes increase (
25).
To date, the PGC has published 24 main articles and 51 secondary analysis articles (see Table S2 in the
online data supplement). At least 141 additional papers have made use of PGC results. Many PGC papers are highly cited, but chief among them is the 2014 schizophrenia report (
15), which ranks among the most highly cited articles in 2014. The PGC is among the leading genomic consortia worldwide for open science and data sharing. These successes are a testimony to the fact-based strategy and persistence of the PGC.
An Update
What have we learned? We now have a sizable body of empirical results relevant to the “common versus rare variant” debate. All common psychiatric disorders with sufficiently large samples have a predominant common disease/common variant contribution (
26–
28). Indeed, this is widely seen across human complex diseases, such as type 2 diabetes mellitus (
29), and anthropometric traits, such as height (
30) and body mass (
31). Demonstrating a major role of common genetic variation in risk for human complex traits (including psychiatric disorders) is so widely and consistently documented that it is no longer particularly newsworthy.
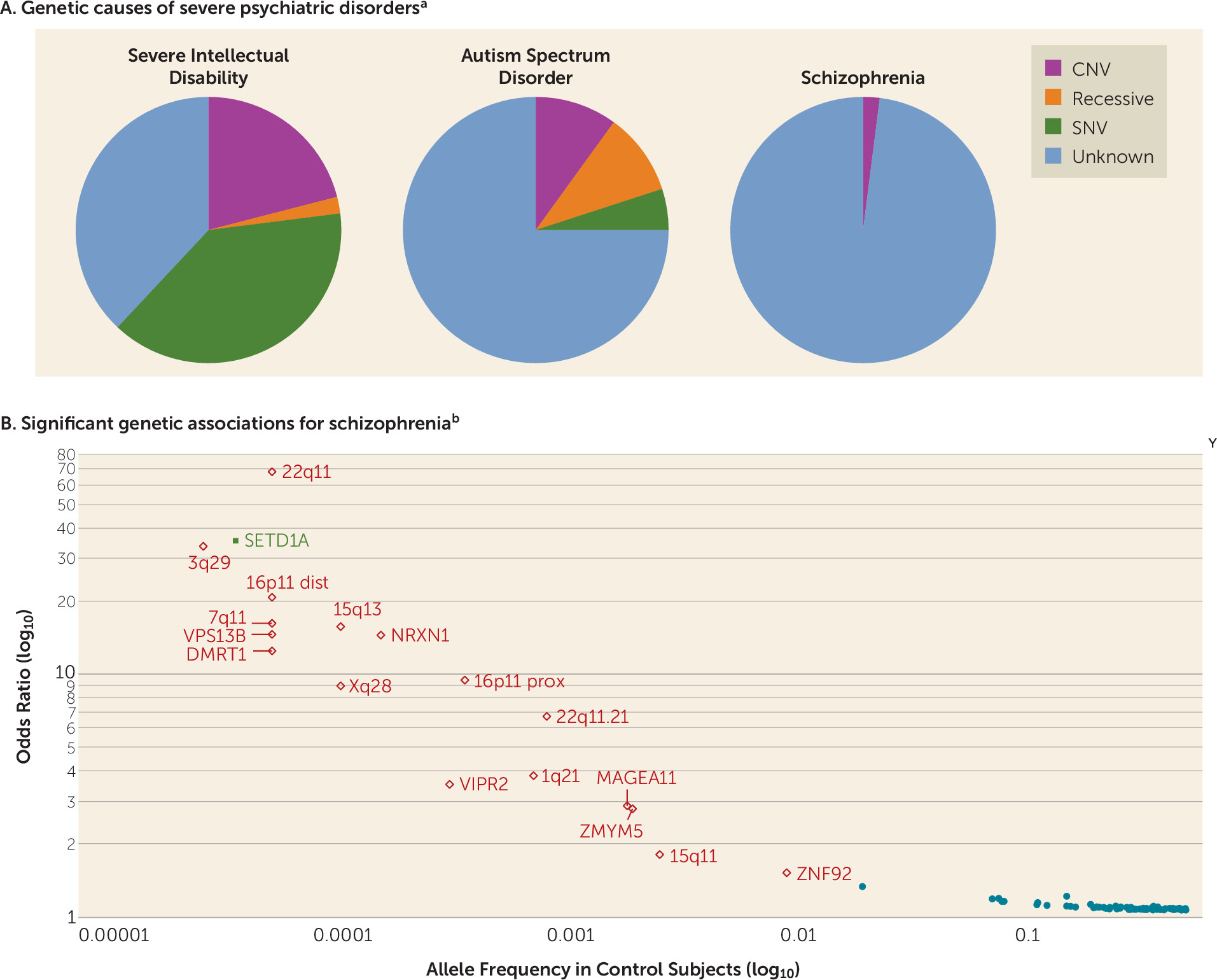
There is a variable contribution of rare variation of strong effect. This tends to be larger for early-onset, severe disorders and lesser for disorders with normal-range developmental trajectories and adult onset (
Figure 2A). However, even for psychiatric disorders with many proven examples of rare variants of strong effect (e.g., intellectual disability or early-onset Alzheimer’s disease), there is always a contribution of common variation. Rare variant studies have proven more difficult than anticipated, because to confidently identify rare variants of strong effect in typical clinical samples requires very large sample sizes, perhaps as many as around 100,000 cases (
38). The protein-coding parts of the genome are replete with inconsequential variation, and current ways to predict functional consequences are imprecise (
39). There is a lot of noise, and the signal is sparser and weaker than anticipated.
Figure 3C shows current sample sizes and notable findings for the PGC working groups. Schizophrenia has accumulated the most data for both common and rare variation.
Figure 2B shows significant results from GWAS, copy number variation (CNV), and exome sequencing studies (
15,
36,
37). Most findings are for common variation. Multiple rare CNVs have been implicated; most are multigenic, and all increase risk for several psychiatric disorders and neurological diseases (
36).
SETD1A is the only gene implicated to date by whole-exome sequencing studies (
37), but other such studies have only found hints of biological pathways by focusing on extremely rare variation (
40,
41). It was widely anticipated that exon variation in the 0.005 to 0.01 allele frequency range would be readily found, but this has not been observed (
42), and a recent study of height required over 700,000 subjects to identify loci in this range (
43). In a direct comparison, common variation had 14–28 times more impact on risk for schizophrenia than rare CNVs or rare exonic variation (
44).
Another major finding has been the repeated empirical documentation of important genetic overlap (particularly common variation) among most or all adult- and childhood-onset psychiatric disorders (
26,
27). It is clear that psychiatric nosology has not “carved nature at the joints.” Moreover, the common variant genetic architecture of many disorders blends into normal phenomena. For example, there are sizable genetic correlations of major depressive disorder with personality traits like neuroticism and readily assessed depressive symptom measures. Other findings suggest that reconceptualizations may be needed. For example, anorexia nervosa had a significant positive genetic correlation with schizophrenia, significant negative genetic correlations with body mass index and unfavorable metabolic measures, and significant positive genetic correlations with favorable metabolic factors. This pattern of findings suggests that the roots of anorexia nervosa may be not only psychiatric, but also metabolic, in origin.
The PGC work group on depression recently completed a report that identified 44 genetic loci for major depressive disorder (
45). This work is notable because of the compressed time scale (2 months from final results to submission) as well as its demonstration of what the findings can tell us. The individual loci yielded multiple strong candidate genes (e.g.,
NEGR1,
RBFOX1, and
SOX5). The findings were associated with clinical features of depression (e.g., earlier age at onset and recurrent and more severe forms of depression). Gene expression patterns in the prefrontal and anterior cingulate cortex most closely matched the genetic findings (these brain regions also show MRI differences between patients with major depression and control subjects). Genes that are targets of antidepressant medications were strongly enriched for depression association signals (p=8.5×10
−10), suggesting pharmacotherapeutic relevance. The genetic bases of lower educational attainment and higher body mass were putatively causal for major depressive disorder, whereas depression and schizophrenia reflected a partly shared biological etiology.
This is an evolving area with regular increases in confident knowledge. To encourage rapid dissemination of results, the PGC regularly compiles and shares a list of the strongest findings for the disorders it studies.
A Consortium Agenda
Attempts to understand the genetic basis of psychiatric disorders—to untangle and concretize the family history risk factor—have never been easy. However, by incorporating empirical results, a data-driven and logical way forward has emerged, and it is likely that these efforts will continue to yield important new knowledge. Many groups are active in this area, but the PGC has emerged as the key umbrella organization for a large portion of this work. A basic description of the PGC and its core principles is given in
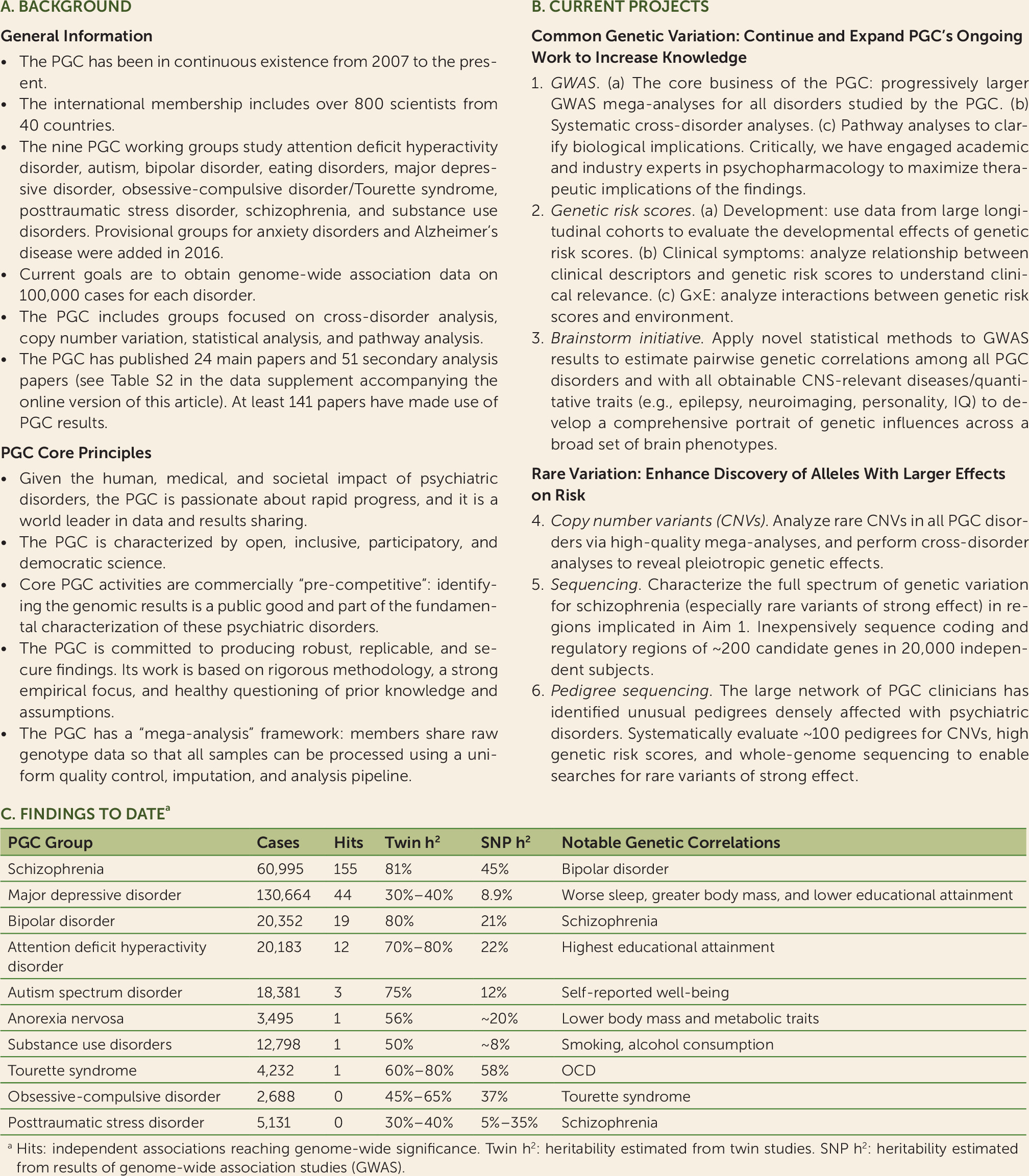
Figure 3A. Key technical aspects include its dedication to rigorous methodologies and its stance as a “mega-analysis” consortium with PGC members sharing individual-level genotype and phenotype data.
With continued support from the National Institute on Mental Health (NIMH) (and new support from the National Institute on Drug Abuse), the PGC recently initiated a program of research designed to deliver “actionable” findings, genomic results that 1) reveal the fundamental biology, 2) inform clinical practice, and 3) deliver new therapeutic targets. This is the central idea of the PGC: to convert the family history risk factor into biologically, clinically, and therapeutically meaningful insights. This program of research has six aims, three focused on common variation and three on rare variation (
Figure 3B).
Aims of Work on Common Genetic Variation
Aim 1
Aim 1 is the core business of the PGC: to conduct progressively larger GWAS mega-analyses and systematic cross-disorder analyses (
46).
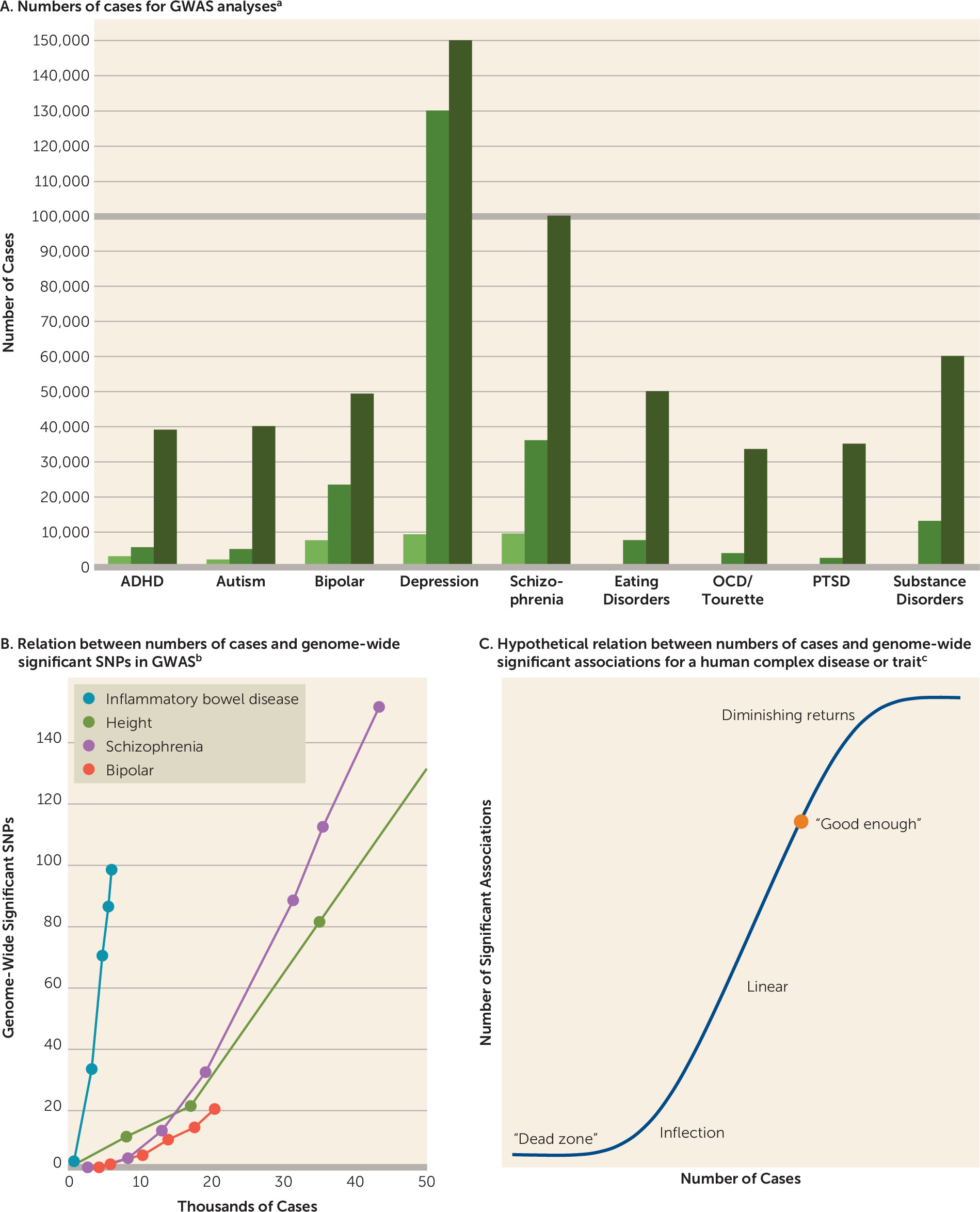
Figure 4A depicts the progression of sample sizes with time. Our goal is for each of the nine disorder working groups to obtain GWAS data on 100,000 cases. More information on case definitions can be found in Table S3 in the
online data supplement.
Figure 4B encapsulates the experience with sample size and numbers of significant associations. Some disorders have a fortuitous architecture; for instance, inflammatory bowel disease obtained a considerable number of findings with relatively small samples. For most other complex traits, the path is slower but with sufficient samples discovery becomes linear.
Figure 4C shows an idealized model of the sigmoid-like discovery process from “dead zone” to asymptote. We suggest that the goal is to get to a “good enough” point, where most genes are identified at least once and the majority of genes in salient biological processes are highlighted. This can provide an etiologic scaffold for studies that use other methods to identify interacting partners in gene networks and pathways that underlie pathogenesis. There may be on the order of 1,000 genes involved in schizophrenia (
47) (for comparison, approximately 13,000 genes are expressed in the brain and about 2,000 at the synapse). Most of the nine PGC disorder working groups have identified at least one genome-wide significant association, several are accumulating moderate numbers of loci, and schizophrenia and major depression appear to be in the linear phase (
Figure 3C).
The PGC has extended its initial efforts in three ways. First, we added four new and highly motivated groups (on eating disorders, obsessive-compulsive disorder/Tourette syndrome, posttraumatic stress disorder [PTSD], and substance use disorders). Provisional groups for anxiety disorders and Alzheimer’s disease have been formed. Second, we hope to markedly increase inclusion of non-European samples (see Figure S1 in the online data supplement). For example, the PGC is now completing a report based on over 12,000 schizophrenia cases from East Asia. The PTSD and substance use disorders groups are studying increasingly larger samples of African Americans. The Stanley Center of the Broad Institute has launched major sample collection efforts for multiple severe psychiatric disorders in Africa, South America, and Asia.
This work is crucial for generalizability. Although it is likely that most (but not all) associations will be observed across the world, there will also be population differences, and it is clear that the application of genetic risk scores globally (see next paragraph) will require risk allele weights derived from the major ancestral populations. Finally, the PGC has engaged academic and industry experts to understand the therapeutic salience of the findings (
48). Indeed, the empirical targets of antipsychotic medications are markedly enriched for the results of schizophrenia GWAS, and this enrichment became clearer with increasing sample sizes, as has the potential pharmacological relevance of calcium channels for psychiatry (
49). The design of rational therapeutics has been an elusive goal for psychiatric indications, and improved genomic knowledge is a precompetitive activity that can make novel drug discovery more efficient (
50).
Aim 2
Aim 2 concerns the analysis of genetic risk scores. For a complex disease or trait, the genetic risk score is a single, normally distributed variable that captures the cumulative effect of risk alleles inherited by an individual (e.g., for schizophrenia, bipolar disorder, or body mass index). Computing a genetic risk score requires a training set (i.e., GWAS results) and genome-wide genotypes for independent test subjects (e.g., a population cohort or participants in a clinical trial). The PGC has made training sets publicly available for multiple disorders. This allows researchers to compute genetic risk scores for whatever use they deem appropriate. These scores are not yet sufficiently discriminating to be useful clinically (
15) but are among the first demonstrably valid biomarkers for psychiatric disorders. Genetic risk scores derived from PGC results have been widely used in psychiatric research for generating patient strata, exploring diagnostic boundaries, identifying cognitive and behavioral correlates of genetic risk that predate clinical disorders, and evaluating the validity of putative cognitive or imaging phenotypes (
51). Many social scientists have embraced the approach, seeing opportunities to study how genetic factors interact with the social environment (e.g., socioeconomic status) to influence health and broader outcomes (
52).
The PGC will systematically evaluate genetic risk scores in three contexts: 1) development—use data from large longitudinal cohorts to evaluate the developmental effects of genetic risk scores; 2) clinical—analyze the relationship between clinical descriptors/symptoms (e.g., early versus late onset, more severe versus milder, or unremitting versus episodic illness) and the genetic risk score to understand clinical relevance; and 3) environment—analyze interactions between the genetic risk score and environmental variables in epidemiological samples.
Aim 3
Aim 3 will use GWAS results to estimate pairwise genetic correlations among all PGC disorders with all obtainable CNS-relevant diseases and quantitative traits (e.g., epilepsy, neuroimaging, personality, and cognition). We will develop a comprehensive portrait of genetic influences across a broad set of brain phenotypes with the intention of improving nosology.
Past epidemiological studies have documented the extensive comorbidities of psychiatric disorders at the phenotype level. Due to limitations inherent to observational studies, understanding whether a phenotypic correlation is potentially causal or if it results from reverse causation or confounding is generally difficult or impossible. Genetic studies now offer complementary strategies. We can readily assess whether a phenotypic association between psychiatric disorders or between a psychiatric disorder and a risk factor is mirrored by a common variant genetic correlation. This can be done using GWAS summary statistics. If the genetic studies are sufficiently large, it is also possible to apply Mendelian randomization to evaluate the potential causality of the association (
53).
For example, a recent PGC study reported sizable positive genetic correlations between major depressive disorder and multiple measures of body mass (
45). We investigated the association by using bidirectional Mendelian randomization and found evidence suggesting a potential genetic causal effect of body mass on risk for depression but not the reverse. These results provide hypotheses for more detailed prospective studies, and the underlying mechanisms are likely to be more complex.
We will operationalize similar analyses for other disorders (e.g., autism with/without intellectual disability, bipolar disorder with/without psychosis or with/without lithium response). Given sex differences in disease prevalence for many disorders, analyses of genetic correlations by sex will be conducted as well. In addition, many of these disorders have significant genetic correlations with cognition, personality, and body mass. Are these genetic correlations putatively causal or due to some other process (confounding or bias)? Differences between disorders will be investigated—for example, body mass has a positive genetic correlation with major depression but a negative genetic correlation with anorexia nervosa (
54).
Aims of Work on Rare Genetic Variation
Aim 4
Aim 4 will continue the PGC’s CNV efforts (
36). The PGC CNV group has created a pipeline to determine the presence or absence of CNVs from the initial intensity files by using multiple algorithms followed by careful quality control and analysis. The initial schizophrenia paper has been published, and this group is now working on bipolar disorder, ADHD, and PTSD and will include more groups with time.
Aim 5
Aim 5 is a “cheap-seq” aim. We will conduct inexpensive (approximately $50/subject) schizophrenia-focused sequencing of 200 genes in 20,000 subjects. Genes will be selected based on all available sequencing results. For 200 genes, we will increase power far more cost-effectively than with whole-exome (10 times cheaper) or whole-genome (25 times cheaper) sequencing in the same time frame. We propose an efficient and affordable way to markedly increase sample sizes for the most promising loci in a new sample of 20,000 subjects.
Aim 6
Aim 6 will systematically evaluate approximately 100 large pedigrees to search for genetic variants of large effect. We have engaged the large network of PGC clinicians in this task. Most experienced clinicians have encountered unusual pedigrees with high concentrations of severe psychiatric disorders. For example, one pedigree has more than 100 individuals with a severe psychiatric disorder, and eight pedigrees have 20 or more affected individuals. Other pedigrees are from genetic isolates in which marriage between relatively close relatives is common. Still other pedigrees have extensive comorbidity with intellectual disability and epilepsy. No one has systematically and comprehensively evaluated a large collection of densely affected pedigrees using comprehensive genomic assays (karyotyping, identity by descent, CNVs, whole genome sequencing, and genetic risk score) combined with a rigorous statistical framework. However, a pedigree very dense with psychiatric disorders can occur because a rare variant of strong effect is segregating in that pedigree or because that pedigree has an unusually high number of common variants of small effect (see reference
55 for an example).
Actionability
Among the aims related to common variants, aim 1 is of biological, clinical, and therapeutic relevance. Aims 2 and 3 are important clinically and for nosology. Of the rare variant aims, all are important biologically and therapeutically (given their potential to identify single genes whose mutational disruption carries high risk).
Issues in the Process of Being Solved
Empirical results from psychiatric genomics have begun to answer many fundamental questions. We point to two major unresolved issues. First, a crucial issue is pinpointing the biological implications of GWAS results. What precise mechanistic hypotheses arise from the findings? If a GWAS “associates” a psychiatric disorder with a specific genomic region, what genes should neuroscientists and molecular biologists study in order to delve more deeply into the basis of a disorder? This is crucial for downstream experimentation, as studying one gene in detail can easily consume several person-years and hundreds of thousands of dollars.
Making connections from DNA sequence variation to a cellular mechanism is sometimes straightforward. This is one reason researchers like to exploit rare exon variants (aims 5 and 6), as the connection to a gene is usually direct and can be logically evaluated with some confidence. Occasionally, common variant findings can be directly implicated; for example, the PGC major depression study found two separate associations on opposite sides of the same gene (
RBFOX1) (
45).
However, such findings are unusual for many common psychiatric disorders (
Figure 2), and connecting the numerous common variant association signals to genes can be challenging.
Figure 5 illustrates typical patterns of results.
Figure 5A shows the
CACNA1C intronic association for schizophrenia; a subsequent study suggested that these variants interact with a regulatory element for
CACNA1C (
56).
Figure 5B depicts the region surrounding
DRD2 (encoding a key target of antipsychotics). This association has been functionally connected to
DRD2 via DNA-DNA regulatory loops (
57).
Figure 5C shows a multigenic region—the association region covers many brain-expressed genes associated with multiple human traits.
Figure 5D depicts a region associated with schizophrenia but far from any known protein-coding gene.
These are typical for GWAS results. Although localization is imprecise, the associated genomic regions are clearly informative as they implicate salient biological pathways (
58), specific genomic features (
59), and targets of common psychiatric medications (
45,
49). Connecting most or all of the findings to specific genes requires additional data based on the function of the human brain, e.g., brain gene expression in brain regions (
60), DNA-DNA looping (
57), and epigenomics (
61). NIMH has funded the PsychENCODE consortium (
61) to conduct an array of functional genomic assays on brain samples from people with severe psychiatric disorders to enable this work. We anticipate considerable progress in this area in the near future.
Second, as discussed above, the genetic basis of most psychiatric disorders shows fundamental connections. For example, the common variant genetic basis of major depressive disorder overlaps significantly with those for anxiety disorders, autism, ADHD, schizophrenia, bipolar disorder, smoking behavior, and anorexia nervosa (
45). Moreover, the presence or absence of some clinical
disorders (major depression, autism, and ADHD) shows strong genetic overlap with the analogous
symptoms in general population samples. Further, the common genetic basis of many psychiatric disorders is often strongly correlated with that of putative subphenotypes (also known as endophenotypes or component phenotypes). For example, the common variant genetic basis of major depression is correlated with that for worse sleep, higher neuroticism, and greater body mass in people without major depression (
45). These results strongly suggest that our diagnostic categories do not define pathophysiological entities. The resolution of these issues will address major unanswered questions: From a genetic perspective, what are these disorders? How are they similar and how are they different?
Common Complaints
Briefly, there are three common complaints about the work of the PGC. First, “The results don’t matter”—the readouts are broad and the effect sizes of individual associated loci are small. In fact, as discussed above, the results are delivering increasingly useful and targeted knowledge (discussed above in the section on aim 1) (
48,
49,
58). The small effect sizes do not constrain the potential utility of targeting the identified genes or pathways—drugs targeting those pathways can have major effects. Small effects can identify “druggable” targets; the canonical example of this is the identification by GWAS of common genetic variation of small effect for multiple cholesterol measures in a gene (
HMGCR) whose protein is the target of a class of cholesterol-lowering medications (
50). Pharmaceutical companies are now following this area closely as genomic data are increasingly crucial to drug development (
50).
Second, “What about unaccounted heritability (h2)?” Heritability estimated from genome-wide single nucleotide polymorphism data (SNP-h2) depends on technical issues and especially sample size. The comparator is estimated from imprecise twin or family data (twin-h2 or pedigree-h2). “Unaccounted h2” refers to the difference between these estimates and attempts to reconcile fundamentally different entities. Still, when the genomic study is sufficiently large (as with schizophrenia), SNP-h2 is around half of the pedigree-h2. A point often missed, however, is that explaining h2 is a minor goal. The main goals of the PGC are to gain biological, clinical, and therapeutic insights, which can arise regardless of the magnitude of heritability accounted for.
Third, because most PGC analyses are based on categorical, case versus control analyses, “PGC cases lack clinical depth.” This was by intention: over 10 years ago (
47), some of us reasoned that fast phenotype characterization that led to affordably large sample sizes was the logical first step (as opposed to large numbers of phenotypes on small numbers of subjects). This was always the first step. The success of this strategy is seen not only in the genome-wide significant loci that we have discovered, but also in the many phenotypes that have been associated with PGC genetic risk score in both clinical and population samples. The second step, under way now, is detailed characterization of genetically informative subsets of cases (e.g., aim 2). In addition, some PGC working groups (e.g., substance use disorders) are currently analyzing quantitative phenotypes.
Conclusions
The PGC is the largest and most systematic genomics effort in the history of psychiatry. In the next 5 years, we propose to markedly scale up our work. By tackling nature as it is and not as we might want it to be, we hope to provide considerable new knowledge about the fundamental basis of psychiatric disorders. Our long-standing commitment to global collaboration, open science, and rapid progress means that we will make our results and tools available in a timely manner. Prediction of the future is always hazardous, but given that we finally have a minimally adequate toolkit for genomics, it is possible that we are entering a golden age of research into the fundamental basis of severe mental illness.
Acknowledgments
The authors thank the investigators of the Psychiatric Genomics Consortium and the hundreds of thousands of people who have shared their life experiences with consortium investigators.