A growing body of literature suggests that major depressive disorder is associated with increased risks of mortality and aging-related phenotypes and diseases, including cardiovascular disease, diabetes, obesity (
1), cancer (
2), cognitive impairment (
3), and frailty (
4). Given the associated negative impact on quality of life and health care costs (
5), it is of interest to investigate whether patients with major depression are prone to accelerated aging.
The literature provides evidence for advanced biological aging in major depressive disorder, as indicated by shorter telomere length (
6,
7) and advanced brain aging (
8). Recently, alternative markers of biological age derived from DNA methylation (also known as “epigenetic clocks”) have been developed. Chronological age can be accurately predicted from methylation data and yields estimates of DNA methylation age (DNAm age) (
9,
10). Furthermore, DNAm age can be studied as either “decelerated” or “accelerated” by regressing it on chronological age to obtain a measure of epigenetic aging. Thus, DNAm age is a promising candidate for reliably investigating accelerated or premature aging in major depression.
Previous studies have shown epigenetic aging in individuals with Down’s syndrome (
11), HIV-positive patients (
12), and individuals with obesity (
13). In addition, epigenetic aging has been associated with poorer physical and cognitive fitness (
14), increased smoking and alcohol use (
15), cancer (
16), Alzheimer’s disease (
17), cardiovascular disease (
16), and an increased risk of mortality (
18). A few studies have investigated DNAm age in relation to schizophrenia (
19,
20), life stress (
21), and posttraumatic stress disorder (
22,
23), with mixed findings. However, studies examining epigenetic aging in relation to major depressive disorder are currently lacking.
In this study, we examined whether major depression is associated with higher epigenetic aging in blood, using a large, clinically well phenotyped sample to further explore associations with clinical characteristics, and we sought to replicate the findings in postmortem brain tissue. We used a sequencing-based approach (
24,
25) that yields almost complete coverage of the CpG methylome, which allowed us to obtain the most accurate DNAm age estimates for our sample and to better explore the biological processes underlying epigenetic aging.
Results
Higher Epigenetic Aging and Major Depression in NESDA
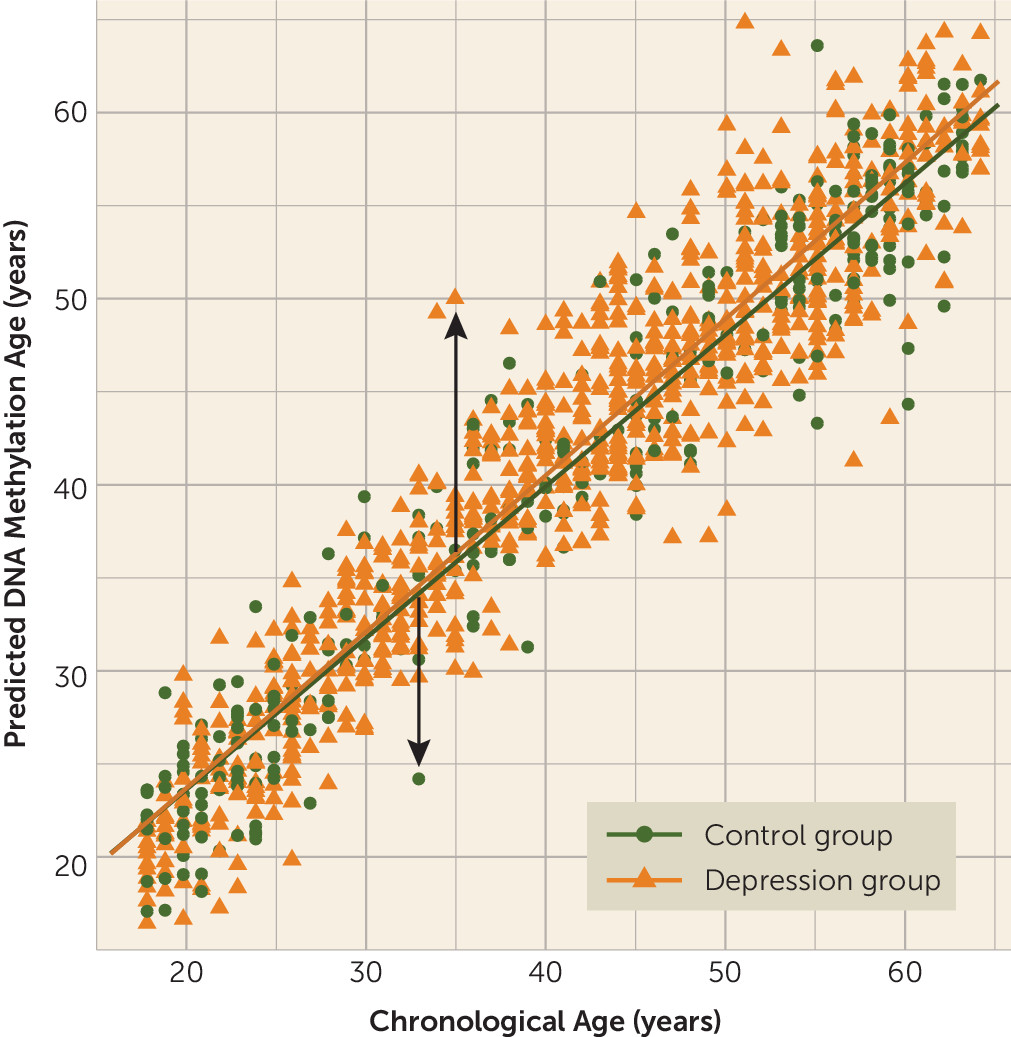
The mean age of the NESDA sample was 41.5 years (SD=13.0, range=18–64), and 64.5% of the sample were female (
Table 1). The depression and control groups did not differ significantly in age, but the depression group had a higher proportion of females (p=0.02) and had fewer years of education on average (p<0.001). As anticipated, patients in the depression group reported higher levels of depression severity and use of antidepressants (all p values, <0.001). The mean childhood trauma score was also higher in the depressed group (p<0.001).
Epigenetic aging showed (by design) a mean of zero (SD=3.58 years), ranging from −13.26 to 15.00. Patients in the depression group had significantly higher epigenetic aging compared with the control group (b=0.64, t=2.65, p=0.008; effect size, Cohen’s d=0.18), indicating that the patient was estimated to be 0.64 years (or 7.68 months) older on average than the control group after full adjustment for covariates (
Table 2). Additional analyses correcting for cell type proportions did not change results and produced a Cohen’s d of 0.14 (see the
data supplement). Consistent with a dose-response effect, a fully adjusted linear regression showed that greater epigenetic aging was significantly associated with higher Inventory of Depressive Symptomatology score in the overall sample (β=0.10, p=0.001). As expected from the high correlation between the DNAm age estimates generated by both models (r=0.98), the above-mentioned results remained unchanged when the same analyses were performed with the DNAm age estimates from the control-subjects-only model (see the
data supplement).
Exploratory Analyses of Epigenetic Aging and Clinical Characteristics
Within the depression groups, we found epigenetic aging to be positively associated with childhood trauma score (β=0.09, p=0.02, see
Table 3). The association between epigenetic aging and Inventory of Depressive Symptomatology score in the overall sample did not remain significant when analyzed only within the depression group (β=0.05, p=0.21), likely because of lower variation in symptom severity. No further significant associations with clinical characteristics were found.
Further analyses revealed that patients in the depression group who had childhood trauma showed the highest epigenetic aging compared with control subjects without childhood trauma (p=0.001, Cohen’s d=0.29), highlighting the fact that this major depression and childhood trauma subgroup is associated with the highest epigenetic aging (see Figure S1 in the data supplement). Notably, greater severity of symptoms of chronic major depression was correlated with childhood trauma (r=0.39, p<0.001), making it difficult to discern which of these two factors drives increased epigenetic aging. Linear regression indeed showed that both childhood trauma (β=0.08, p=0.01) and depression severity score (β=0.07, p=0.03) were significant predictors of epigenetic aging when analyzed in the same model.
Replication in Postmortem Brain Samples
The mean age of the postmortem brain samples was 55.2 years (SD=19.3, range=20–100), and 45.4% of the sample were female. Groups were matched on age and sex. The mean postmortem interval was 35.1 hours (SD=21.1), and the mean pH was 6.51 (SD=0.25) across samples. Epigenetic aging was uncorrelated with pH or postmortem interval.
Table 4 summarizes the descriptive characteristics by brain collection. Only the control (N=67) and depression (N=74) samples from the same brain collection were included in analyses (see the
data supplement for more detail).
Our replication findings in independent brain samples supported our findings in NESDA and again showed that the epigenetic aging was higher in the depression group than in the control group (b=1.11, χ2=3.41, p=0.03). The beta indicates that the depression group was estimated to be on average 1.11 years older than the control group. The phenotype information available from the postmortem samples was limited, and therefore we were unable to attempt any replication of the exploratory clinical associations observed, such as childhood trauma, in NESDA.
Enrichment Testing and Gene Ontology Analyses
When evaluating the overlap between both epigenetic aging indicators, we found that after correcting for multiple testing, the top 1% findings from the epigenetic aging MWAS in blood were significantly enriched for CpGs in the top 0.5% of the epigenetic aging MWAS from brain (odds ratio=1.19, p<0.001). To examine possible processes underlying epigenetic aging in both tissues, we performed pathway analyses on the 1,084 overlapping CpGs associated with epigenetic aging, leading to 330 genes (90.7%) that were present in at least one GO category. Subsequently, this resulted in 53 significantly enriched GO terms (see Table S4 in the data supplement). The top GO terms included neurogenesis (p=9.79×10−9), neuron differentiation (p=5.34×10−8), and regulation of neuron death (p=4.67×10−5), indicating that several depression-relevant pathways were enriched in the cross-tissue epigenetic aging indicators.
Discussion
To the best of our knowledge, this is the first time higher epigenetic aging in patients with major depression compared with control subjects has been demonstrated. Exploratory analyses suggested even more pronounced epigenetic aging in patients with major depression who had higher childhood trauma scores. The case-control difference in blood was replicated in postmortem brain tissue. Finally, analyses showed significantly enriched neuronal pathways associated with the overlap between epigenetic aging–associated CpGs from blood and brain tissue.
Replication of our main finding in postmortem brain tissue bolstered confidence in the observed higher epigenetic aging in major depression. Moreover, the significantly enriched overlap suggests that at least some processes affecting epigenetic aging are at play in both blood and brain. There is some evidence that blood and brain show concordance in methylation (
40) and epigenetic aging (
10). However, considering the interactions between stress, central and peripheral immune processes, and neurobiology (
41), it is plausible and likely that epigenetic aging in major depression is also dictated by many systemic processes. Nonetheless, more work is needed to confirm the higher epigenetic aging findings and to better characterize advanced aging–associated genes and their implications in major depression.
DNAm age is just one of the several available markers of biological aging (
35). The present study confirms advanced or premature biological aging in major depression with a novel platform and is consistent with the literature regarding telomere length as biological marker of aging in major depression (
7,
42). Also in line with other studies (
43,
44), post hoc analyses between telomere length and epigenetic aging showed nonsignificant relationships, suggesting that both measures likely independently track different aspects of biological aging. Similarly, other post hoc analyses showed that telomere length did not alter our findings when accounted for, providing further evidence that epigenetic aging captures significant aging signal different from telomere length (see the
data supplement).
We found that childhood trauma was positively associated with higher epigenetic aging in patients with major depression. It seems conceivable that major depression and accumulated stress throughout the lifetime due to childhood trauma may alter the epigenetic landscape and influence genomic regulation and function (
45). However, this study did not identify additional relationships between higher epigenetic aging and more cumulative clinical characteristics, such as earlier onset age or longer duration of depression. Rather, our findings suggest that higher epigenetic aging in major depression may be driven largely by severity of illness.
Alternatively, childhood trauma may produce long-lasting epigenetic “scars” that have an impact on major depression and advanced or premature aging processes later in life. Individuals with childhood trauma and depressive disorders have an earlier onset age, higher symptom severity, more comorbidities, increased suicidality, and poorer treatment response than patients without childhood trauma (
46,
47). As Teicher and Samson (
47) have suggested, presence of childhood trauma is associated with a clinically and neurobiologically distinct subtype of depression.
Strikingly, three out of 10 top GO categories enriched across tissues included neuronal pathways. Epigenetic mechanisms are critical in early brain development, adult neurogenesis, and late-stage brain maturation (
48). Given that these are all processes that seem markedly aberrant in major depression (
49), the implicated pathways suggest that epigenetic aging in major depression directly contributes to symptomology. Additionally, the degree of overlap between the top 1% findings of epigenetic aging from blood and brain and the top 0.5% findings of chronological age in NESDA (odds ratio=85.31, p<0.001, and odds ratio=1.64, p<0.001) was highly significant, suggesting that biological aging overlaps with the same epigenetic processes that underlie chronological aging.
Although effect sizes such as those we observed here are common in depression research (e.g., oxidative stress, brain-derived neurotrophic factor, and cortisol yield effect sizes ranging from 0.15 to 0.31 [
50–
52]), it is possible that this is an underestimate. The reason is that DNAm age is estimated from the residuals of the regression of methylation data on chronological age. This residual variance comprises two components: true unique variance associated with DNAm age, and measurement errors. However, because the residual variance is very small (the correlation between chronological age and methylation data was 0.95), even small measurement errors can have a large negative effect on the reliability of the DNAm age that is defined as reliability=Var(DNAm age)/(Var[DNAm age]+Var[errors of measurement]). As a less than perfect reliability will attenuate the correlation of DNAm age and depression status, the real effect size may have been underestimated.
Strengths of this study are the replication in postmortem brain tissue and inclusion of a large, clinically well characterized, and representative sample, with data that included many potential confounders that did not explain our findings. Given the full methylome coverage, we were also able to examine which biological pathways seemingly underlie epigenetic aging. However, our findings should also be considered against some limitations. A direct comparison of our MBD-seq-based epigenetic clock and existing Illumina array-based clocks was not possible and was beyond the scope of this study. However, a side-by-side comparison is an interesting endeavor for a future methodological study. Furthermore, with the present cross-sectional data, we were not able to disentangle whether greater epigenetic aging in major depression truly reflects aging acceleration over time or whether some individuals have increased epigenetic aging from birth or before adulthood that continues to be stable thereafter (
53). Further studies with longitudinal designs are needed to distinguish the two possibilities.
In conclusion, our findings show that DNAm age from both blood and brain of patients with major depression is higher than their corresponding chronological age, which may contribute to their increased risk for mortality and aging-related diseases. Furthermore, higher childhood trauma scores correlated with higher epigenetic aging in patients with major depression. Taken together, our findings suggest that higher methylation aging in major depression is present in both blood and brain and that higher epigenetic aging largely overlaps with the same underpinnings associated with chronological aging. Further research is needed to investigate the causal relationships between age-associated alterations in DNA methylation and major depression.