“Mr. A,” a 26-year-old man admitted to the early psychosis inpatient unit presented in an acute manic state with impulsivity, disinhibition, overfamiliarity, pressure of speech, flight of ideas, restlessness, reduced sleep, and grandiose and paranoid delusions associated with visual and auditory hallucinations as well as ideas of reference. During the week before admission, Mr. A had been spending a lot of money, consuming alcohol and marijuana on a daily basis, and been irritable and aggressive toward others. His Positive and Negative Syndrome Scale (PANSS) score was 103 at admission. Extensive examination of neurocognitive functions was not possible because of the language barrier. The patient had suffered one past episode of mania 2 years earlier, but no depressive episodes. Physical and neurological examinations were unremarkable. The patient appeared well nourished, with a BMI of 20. Apart from slightly decreased folic acid levels (9.5 nmol/L; normal range, 9.53–44.9 nmol/L), blood results at admission were normal (hematologic parameters, coagulatory parameters [INR 1.0], electrolytes, creatinine [0.58 mg/dL], bilirubin [total 0.26 mg/dL, direct 0.16 mg/dL, indirect 0.10 mg/dL], albumin [42.3 g/L], amylase, lipase, cholinesterase, AST [27 U/L], ALT [30 U/L], GGT [17 U/L], C-reactive protein, serum ferritin, vitamin B12, thyroid-stimulating hormone, 1,25-dihydroxycholecalciferol, and carbohydrate-deficient transferrin as a marker of recent alcohol abuse). A urine drug screen was positive for cannabinoids, and a brain MRI scan did not reveal any structural abnormalities.
Pharmacotherapy was initiated with valproic acid at 500 mg/day, risperidone at 1 mg/day, and lorazepam at 2.5 mg/day, and the dosages were increased expeditiously. On day 7 of treatment, Mr. A’s manic symptoms—particularly agitation and irritability—remained unchanged, and his plasma levels of valproic acid (1200 mg/day) and risperidone (6 mg/day) were below therapeutic range. Consequently, the dosages were increased in a stepwise manner (valproic acid 2500 mg/day, risperidone 7.5 mg/day, lorazepam 7.5 mg/day). On day 14, the patient’s plasma levels of valproic acid (2500 mg/day) and risperidone (8 mg/day) were within therapeutic range, and a slight improvement was observed in his psychopathology, especially with respect to impulsivity, disinhibition, and overfamiliarity. Starting at day 15, Mr. A complained of daytime sedation, and consequently the lorazepam dosage was decreased. On day 16, the patient still appeared sedated, and over a period of 2–3 hours, he became progressively less alert and exhibited psychomotor slowing.
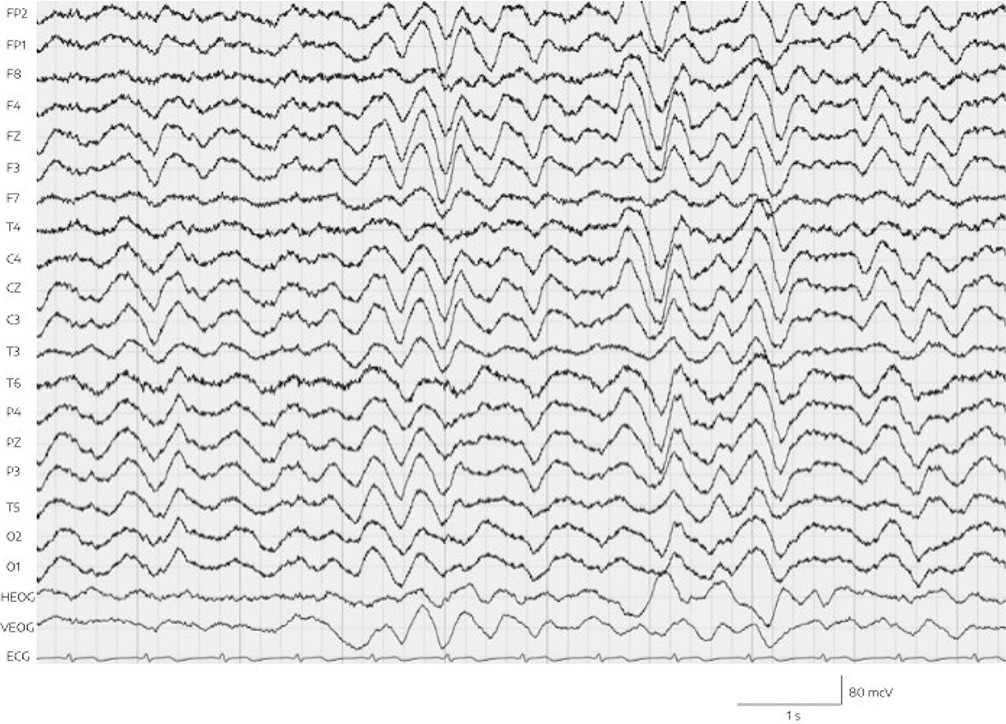
EEG was performed the same day, and showed pathologic alterations. Specifically, continuous generalized delta wave activity (with a frequency of 2–3 Hz and an amplitude of approximately 50 to 60 mV) with intermittent generalized increases (to approximately 130 mV and without adequate reaction to external stimuli) was observed, but no epileptiform abnormalities and no triphasic waves (Figure 1). Standard hematologic, coagulation, renal, and metabolic parameters were normal. Hepatic enzymes were within the normal range. The patient’s serum ammonia level, however, had increased 10-fold to 412.2 μmol/L (normal range, 14.7–55.3 μmol/L). Furthermore, the valproic acid plasma level (1024 μmol/L) was also clearly above the therapeutic range of 350–700 μmol/L.
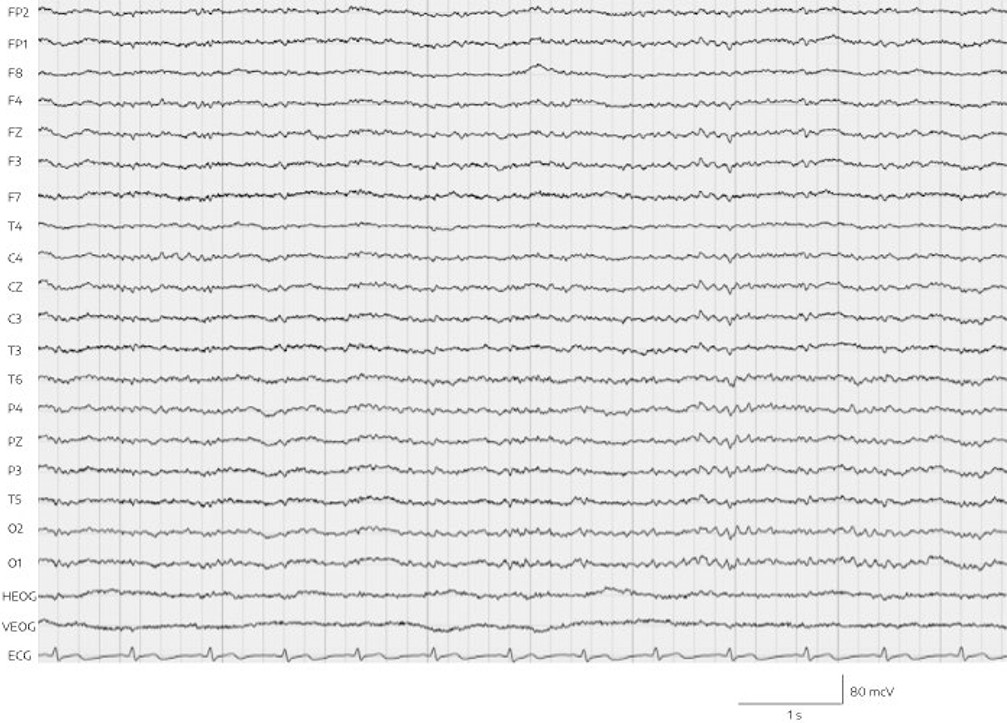
The combination of significantly increased ammonia and increased valproic acid levels, with clinical signs of encephalopathy such as deep sedation and motor retardation as well as an aberrant EEG, led us to consider a valproic acid–associated hyperammonemic encephalopathy. For closer observation and regular monitoring of vital signs, Mr. A was transferred to a psychiatric intermediate care unit. Valproic acid was discontinued at once, and forced diuresis with 2000 mL isotonic fluid and 40 g l-ornithine-l-aspartate to support urea synthesis and detoxification of ammonia was performed every 12 hours. The next day, the patient was alert and awake. His ammonia levels had returned to normal (51.7 μmol/L) within 24 hours, with valproic acid initially at therapeutic levels (525 μmol/L) and 3 days later at 29 μmol/L. Four days after the discontinuation of valproic acid, a follow-up EEG demonstrated a normal alpha rhythm without generalized abnormalities (Figure 2). The patient did not suffer any permanent neurological damage, and he returned to our standard psychiatric ward within 6 days. In order to replace valproic acid, quetiapine was started and titrated to 600 mg/day alongside risperidone and lorazepam. Subsequently, orally administered risperidone was switched to intramuscular depot medication of paliperidone.
Mr. A was discharged several weeks later in full remission with no manic or psychotic symptoms (his PANSS score at discharge was 31). To test for the presence of any urea cycle defects, a laboratory screening of plasma and urine organic acids was performed 4 months after the event. Genetic testing for polymorphisms of the cytochrome P450 enzymes revealed a heterozygous mutation and probable phenotype of intermediate metabolizer in CYP2C19 (genotype: *1/*2) and extensive metabolizer for CYP2C9 (genotype: *1/*1), CYP2D6 (genotype: *1/*41), CYP3A4 (genotype: *1/*1), and CYP3A5 (genotype: *3/*3).
FIGURE 1. Delta EEG in both hemispheres and ECG lead (bottom) on day 16 of admissiona
Valproate in its various forms (valproic acid [sodium valproate] and valproate semisodium), originally developed as an antiepileptic drug, is a first-line therapy for the treatment of acute mania, with recommendation grade 1 and clinical evidence A, except in younger women (recommendation grade “2”), according to current guidelines (1). Valproate acts by increasing gamma-aminobutyric acid (GABA) levels via the inhibition of GABA-metabolizing enzymes and GABA synthesis catalysis, and further by inhibiting N-methyl-d-aspartate (NMDA) receptors and neuronal sodium and calcium channels (2). Interferences in cell metabolism and structure of lipid membranes have been described. Some of the most common side effects of valproate treatment include tremor, confusion, anemia, thrombocytopenia, nausea, and weight gain. Severe adverse events such as pancreatitis, hepatotoxicity, coagulatory defects, and potential teratogenicity (associated with exposure during pregnancy) also may occur with valproate treatment (3). Notably, chronic therapy with valproate can occasionally produce a syndrome of cognitive deterioration and/or parkinsonism, sometimes accompanied by ataxia and/or hearing loss, which is reversible after valproate is stopped but advances if valproate is continued. Cortical pseudoatrophy and enlargement of the lateral ventricles are typical radiologic signs of this side effect, which can occur weeks to years into treatment with valproate (4). An incidence between 1% and 2% was reported in a cohort of epilepsy patients under valproate treatment (5).
Valproate-associated encephalopathy is described as a rare but serious adverse event, predominantly reported in young children with epilepsy who have innate metabolic disorders (6). Combinations of anticonvulsant drugs seem to increase the risk of encephalopathy (6). Valproate-associated encephalopathy is an acute and critical condition characterized by reduced consciousness, focal neurological deficits, vomiting, vertigo, lethargy, and cognitive slowing, often with hyperammonemia and sometimes with hepatopathy (3). However, hyperammonemia and hepatopathy do not occur in all patients (3, 7). Indeed, in a study of patients with epilepsy, different phenotypes of valproate-associated encephalopathy could be identified, and different pathogenic pathways were suspected as the explanation for this heterogeneity (3).
Data on incidence rates of valproate encephalopathy in epilepsy patients are inconclusive, and some cases may be overlooked (3). In psychiatric patients, 25 cases of valproate-associated encephalopathy were reported between 1980 and 2017 (7–15). However, there is evidence for broader relevance, as a recent cross-sectional study (16) found a rate of 2.5% for valproate-induced hyperammonemic encephalopathy in a general hospital population with at least one psychiatric disorder. Possibly, in some cases, signs and symptoms of encephalopathy may go unnoticed because of heterogeneity in clinical presentation and laboratory findings.
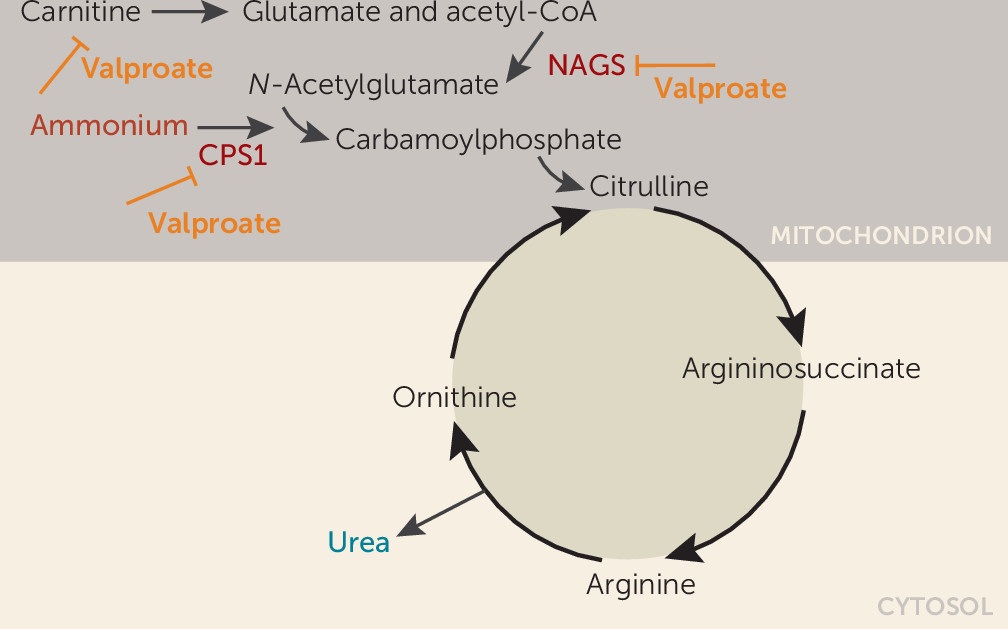
Our patient developed a fulminant onset of encephalopathy and hyperammonemia associated with antimanic treatment with valproic acid. Interestingly, he showed clinical improvement and tolerated the medication well for 2 weeks with plasma levels within normal ranges until a sudden onset of clinical signs. The acute clinical onset and the 10-fold elevation of ammonia, while valproate had initially been within a therapeutic range, could suggest an underlying genetic vulnerability in the patient. In fact, direct inhibition of the urea cycle by valproate has been suggested as the primary mechanism in patients with isolated hyperammonemic encephalopathy without hepatopathy (3). Valproate inhibition of ammonia elimination in the urea cycle is mediated by the inhibition of N-acetylglutamate synthase (NAGS) and carbamylphosphate synthase 1 (CPS1) activity, and by the interference in carnitine transport of acyl-coenzyme A via the mitochondrial matrix (Figure 3) (17). Polymorphisms in CPS1 and NAGS genes have been associated with hyperammonemia in valproate treatment (18).
FIGURE 3. Schematic depiction of valproate inhibition of ammonia elimination in the urea cyclea
A screening for innate defects of urea cycle was negative in our patient; plasma glutamic acid was only slightly increased to 87.8 μmol/L (normal range, 1–57 μmol/L), and there were no signs of orotic acid in the urine. Furthermore, no anamnestic hints were found in the patient’s own and in his family history (e.g., no unexplained deaths of young male children), and his ammonia levels were normal in the absence of valproate. Hence, severe innate urea cycle defects (Figure 4) can be excluded as the mechanism of action in our patient. However, in the plasma amino acid screening, months after the event, the patient’s serum showed hyperprolinemia and elevated leucine and valine levels as markers of ketosis. Alanine and glycine plasma levels were also elevated to a medium extent, indicating a secondary rather than a primary mitochondrial disease, that is, with an environmental etiology rather than a genetic etiology (nuclear DNA/mitochondrial DNA). As valproate depletes mitochondrial acetyl-CoA (Figure 3), an inborn mitochondriopathy, demasked by valproate, could also be a relevant mechanism for hyperammonemia in our patient. However, with the diagnostic tests described above, a definite diagnosis cannot be made yet. Further genetic testing at a specialized center for inborn errors of metabolism was recommended. Figure 4 lists inborn errors of metabolism that favor the occurrence of hyperammonemic encephalopathy under valproate treatment. A review by Segura-Bruna et al. (19) offers an extensive discussion on pathophysiology and a broad differential diagnostic list of factors predisposing to hyperammonemia.
FIGURE 4. Inborn errors of metabolism as risk factors for hyperammonemic encephalopathy under valproate treatmenta
In our patient, signs of encephalopathy were obvious and swiftly led to the right diagnostic and therapeutic approach. In many cases, however, clinical signs may not present in such a clear manner. New onset of neurologic symptoms during valproate treatment should lead to diagnostic measures such as examination of ammonia levels and EEG, even when valproate levels and liver enzymes are within normal ranges. Nonetheless, we do not recommend routine measurements of ammonia in all patients under valproate treatment; in a 5-year single-center observational study (N=158) (20), hyperammonemia was observed in 20%−30% of epilepsy patients on valproate and was dose dependent. The risk of ammonia elevation was further increased when valproate was combined with liver enzyme–inducing antiepileptic or antipsychotic drugs. In an acutely ill psychiatric intensive care unit population, hyperammonemia was observed in every second patient on valproate, and also in every fifth patient on another mood stabilizer, without the occurrence of valproate-associated encephalopathy (21).
In conclusion, valproate-associated encephalopathy remains a possibly underestimated and heterogeneous entity with a variety of mechanisms compatible with the broad therapeutic spectrum of valproic acid. Based on the reported case, we recommend close monitoring of newly developed neurologic symptoms such as cognitive deterioration, ataxia, tremor, confusion, and changes in consciousness during valproate treatment, even if the medication was not started recently. If valproate-associated encephalopathy is considered, tests of serum ammonia levels and EEG should be performed immediately. Mild cases of encephalopathy, characterized mostly by confusion, may be easily overlooked.
Acknowledgments
The authors thank Alexander Kaltenboeck and Tarek Zghoul for their help in editing the manuscript.
References
1.
Grunze H, Vieta E, Goodwin GM, et al: The World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the biological treatment of bipolar disorders: update 2009 on the treatment of acute mania. World J Biol Psychiatry 2009; 10:85–116
Gerstner T, Buesing D, Longin E, et al: Valproic acid induced encephalopathy: 19 new cases in Germany from 1994 to 2003: a side effect associated to VPA-therapy not only in young children. Seizure 2006; 15:443–448
Ristić AJ, Vojvodić N, Janković S, et al: The frequency of reversible parkinsonism and cognitive decline associated with valproate treatment: a study of 364 patients with different types of epilepsy. Epilepsia 2006; 47:2183–2185
Al-sharefi A, Bilous R: Reversible weakness and encephalopathy while on long-term valproate treatment due to carnitine deficiency. BMJ Case Rep 2015; 2015:bcr2015210727
Dawson LP, Lee SJ, Hollander YS: Valproate-induced hyperammonemic encephalopathy associated with urinary tract infection and urinary retention in the psychiatric setting. Aust N Z J Psychiatry 2016; 50:1110
Elwadhi D, Prakash R, Gupta M: The menacing side of valproate: a case series of valproate-induced hyperammonemia. Indian J Psychol Med 2017; 39:668–670
Patel N, Landry KB, Fargason RE, et al: Reversible encephalopathy due to valproic acid induced hyperammonemia in a patient with bipolar I disorder: a cautionary report. Psychopharmacol Bull 2017; 47:40–44
Lewis C, Tesar GE, Dale R: Valproate-induced hyperammonemic encephalopathy in general hospital patients with one or more psychiatric disorders. Psychosomatics 2017; 58:415–420
Aires CCP, van Cruchten A, Ijlst L, et al: New insights on the mechanisms of valproate-induced hyperammonemia: inhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J Hepatol 2011; 55:426–434
Tseng Y-L, Huang C-R, Lin C-H, et al: Risk factors of hyperammonemia in patients with epilepsy under valproic acid therapy. Medicine (Baltimore) 2014; 93:e66
The Department of Psychiatry and Psychotherapy, Clinical Division of Social Psychiatry (Baumgartner, Hoeflich, Hinterbuchinger, Fellinger, Friedrich, Mossaheb), and the Department of Psychiatry and Psychotherapy, Clinical Division of General Psychiatry (Graf, Frey), Medical University of Vienna.
The Department of Psychiatry and Psychotherapy, Clinical Division of Social Psychiatry (Baumgartner, Hoeflich, Hinterbuchinger, Fellinger, Friedrich, Mossaheb), and the Department of Psychiatry and Psychotherapy, Clinical Division of General Psychiatry (Graf, Frey), Medical University of Vienna.
The Department of Psychiatry and Psychotherapy, Clinical Division of Social Psychiatry (Baumgartner, Hoeflich, Hinterbuchinger, Fellinger, Friedrich, Mossaheb), and the Department of Psychiatry and Psychotherapy, Clinical Division of General Psychiatry (Graf, Frey), Medical University of Vienna.
The Department of Psychiatry and Psychotherapy, Clinical Division of Social Psychiatry (Baumgartner, Hoeflich, Hinterbuchinger, Fellinger, Friedrich, Mossaheb), and the Department of Psychiatry and Psychotherapy, Clinical Division of General Psychiatry (Graf, Frey), Medical University of Vienna.
The Department of Psychiatry and Psychotherapy, Clinical Division of Social Psychiatry (Baumgartner, Hoeflich, Hinterbuchinger, Fellinger, Friedrich, Mossaheb), and the Department of Psychiatry and Psychotherapy, Clinical Division of General Psychiatry (Graf, Frey), Medical University of Vienna.
The Department of Psychiatry and Psychotherapy, Clinical Division of Social Psychiatry (Baumgartner, Hoeflich, Hinterbuchinger, Fellinger, Friedrich, Mossaheb), and the Department of Psychiatry and Psychotherapy, Clinical Division of General Psychiatry (Graf, Frey), Medical University of Vienna.
The Department of Psychiatry and Psychotherapy, Clinical Division of Social Psychiatry (Baumgartner, Hoeflich, Hinterbuchinger, Fellinger, Friedrich, Mossaheb), and the Department of Psychiatry and Psychotherapy, Clinical Division of General Psychiatry (Graf, Frey), Medical University of Vienna.
The Department of Psychiatry and Psychotherapy, Clinical Division of Social Psychiatry (Baumgartner, Hoeflich, Hinterbuchinger, Fellinger, Friedrich, Mossaheb), and the Department of Psychiatry and Psychotherapy, Clinical Division of General Psychiatry (Graf, Frey), Medical University of Vienna.
The authors report no financial relationships with commercial interests.
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).
If the address matches an existing account you will receive an email with instructions to retrieve your username
Create a new account
Change Password
Password Changed Successfully
Your password has been changed
Login
Reset password
Can't sign in? Forgot your password?
Enter your email address below and we will send you the reset instructions
If the address matches an existing account you will receive an email with instructions to reset your password.
Change Password
Congrats!
Your Phone has been verified
×
As described within the American Psychiatric Association (APA)'s Privacy Policy and Terms of Use, this website utilizes cookies, including for the purpose of offering an optimal online experience and services tailored to your preferences. Please read the entire Privacy Policy and Terms of Use. By closing this message, browsing this website, continuing the navigation, or otherwise continuing to use the APA's websites, you confirm that you understand and accept the terms of the Privacy Policy and Terms of Use, including the utilization of cookies.