Schizophrenia is a complex neuropsychiatric syndrome with a heterogeneous genetic, neurobiological, and phenotypic profile. Currently, no objective biological measures—that is, biomarkers—are available that inform diagnostic or treatment decisions. A biomarker, as outlined by the U.S. Food and Drug Administration and National Institutes of Health Biomarker Working Group, is “a characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions” (
1). Neuroimaging is a strong candidate for biomarker development in schizophrenia. Imaging can capture phenotypic variations in molecular and cellular disease targets or in brain circuits that are a unique representation of gene-environment interactions and are associated with behavioral alterations (
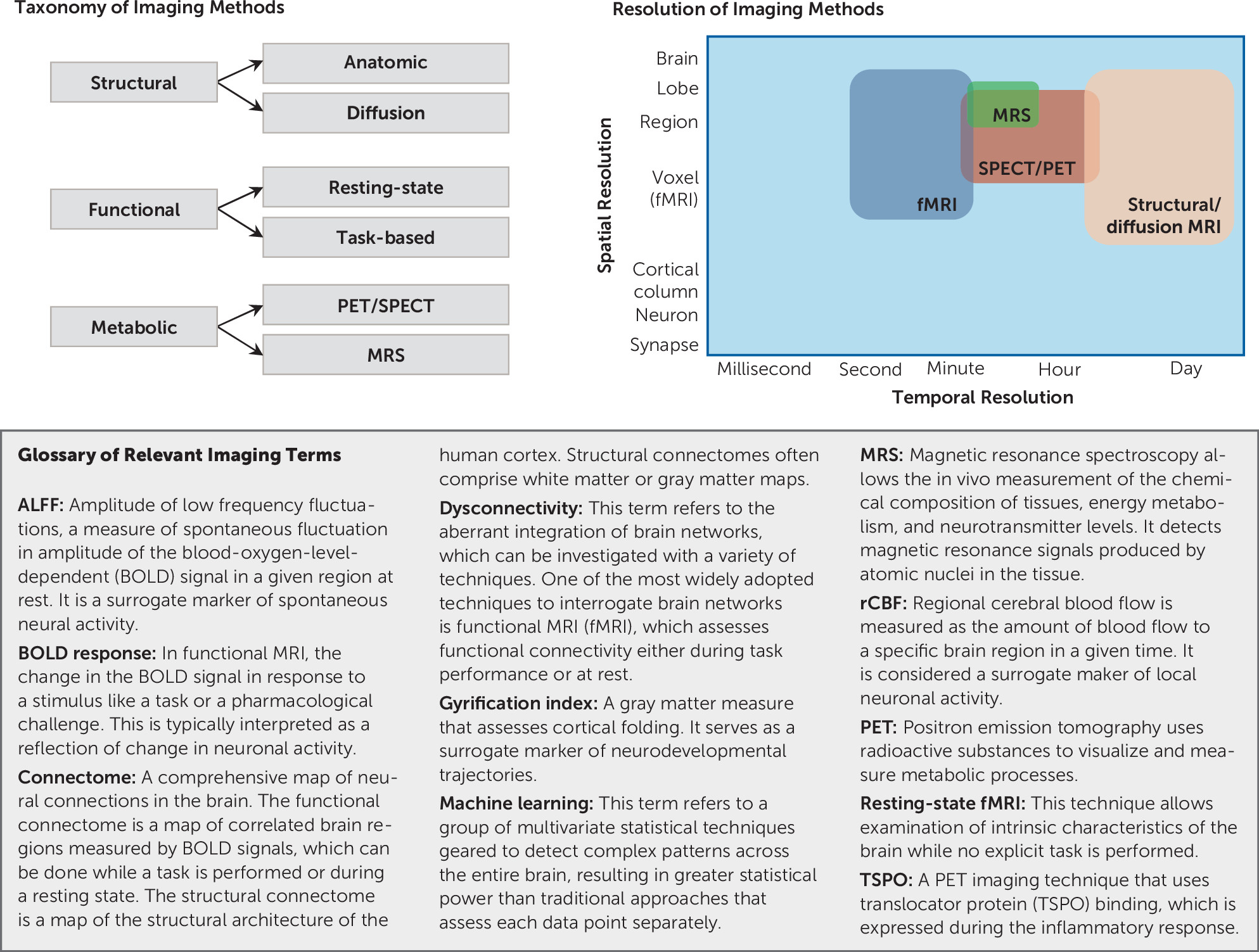
2). It offers versatility in terms of measuring multiple pathophysiological mechanisms, including brain structural integrity deficits, functional dysconnectivity, and altered neurotransmitter systems (
Figure 1) (
3). For a biomarker to be practically useful, it must be a proxy of a clinically relevant measure. It needs to have an acceptable level of sensitivity, specificity, and predictive value (
4). Ideally, it will also be easily quantifiable and cost-effective.
Mechanistically Plausible Targets for Biomarker Development in Schizophrenia
Mechanistically based biomarkers represent a direct measure of the pathophysiological underpinnings of the disease process (
6) and thus can serve as true intermediate or surrogate endpoints (
Figure 2). These biomarkers can validate new treatment targets or pathways, predict treatment response, aid in selection of patients for therapy, determine therapeutic regimens, and provide a rationale for personalized treatments (
7). Ideally, biomarker development targets would reflect fundamental neurobiological alterations, have analogues in preclinical models, correlate with measures of clinical symptom severity, and be consistent with models of disease pathology (
8). Here, we selectively review mechanistically plausible biomarker development targets that meet these criteria (
Table 1).
Dopamine hyperactivity has a long history as a prominent pathophysiologic hypothesis of schizophrenia (
9,
10), as medications that treat psychosis are dopamine D
2 receptor antagonists (
11) and dopamine-enhancing agents such as stimulants are psychotomimetic (
12). In rodent models, amphetamine administration induces locomotor sensitization that is accompanied by an increase in dopamine efflux from the nucleus accumbens and dorsal striatum (
13). In schizophrenia patients, a link between dopamine dysregulation and psychosis severity (
14) and a relationship between baseline dopamine D
2 receptor occupancy and antipsychotic treatment response have been reported (
15–
17).
N-Methyl-
d-aspartate receptor (NMDAR) hypofunction is widely hypothesized to be a central neurobiological alteration in schizophrenia (
18,
19). Experimental evidence supports NMDAR hypofunction as a high-priority target for biomarker development. NMDAR hypofunction on the γ-aminobutyric acid (GABAergic) interneuron causes disinhibition of the glutamatergic pyramidal cell (
20–
22). The presence of a hyperglutamatergic state in different brain areas in patients with schizophrenia has been empirically confirmed and replicated in a number of magnetic resonance spectroscopy (MRS) studies (
23–
29). Preclinical data further support this target by demonstrating that experimentally induced NMDAR hypofunction results in increased firing of glutamatergic neurons in animal models (
30) and produces psychosis-like behavioral phenotypes (
31–
33) and glutamatergic excess in healthy human subjects (
34–
36). Unfortunately, no validated positron emission tomography (PET) ligand visualizing NMDAR function in vivo is available to date (
37).
Another biomarker development target is hippocampal hyperactivity. Here, the model suggests that a hyperglutamatergic state causes hippocampal hyperactivity, which then may result in downstream dopamine circuit dysregulation and psychotic symptoms (
38,
39). Several studies reported hippocampal hyperactivity in patients with schizophrenia (
40–
43) and found a relationship between hippocampal regional cerebral blood flow and psychosis symptom severity (
44) as well as antipsychotic treatment response (
45). Linking cellular-level mechanisms and neuroimaging findings, Schobel and colleagues (
46) reported a series of experiments in a preclinical model of psychosis showing that ketamine administration causes an increase in extracellular glutamate and hippocampal hyperactivity. Experiments in a methylazoxymethanol acetate model, which recapitulates a developmental disruption leading to neurophysiological and behavioral deficits that resemble components of schizophrenia, further demonstrate that hippocampal hyperactivity results in increased dopaminergic signaling, which can be reversed by inactivating the ventral hippocampus (
47), suggesting possible translational utility of this marker.
Neuroinflammation or immune dysregulation as plausible biomarkers for schizophrenia are rooted in the observation of a link between autoimmune processes and development of psychosis (
48). In this model, microglia are primed during early development into a hyperresponsive mode, then shift to a proinflammatory state in response to stress during critical developmental periods. This in turn can result in aberrant neurotransmission, synaptic pruning, and structural injury of neurons and glia (
49). Maternal immune activation models in rodents show a postpubertal symptom onset and have structural, neurochemical, and behavioral abnormalities recapitulating the human clinical picture (
50). Evidence of immune dysregulation has also been reported in postmortem studies (
51) and in studies examining cytokines in cerebrospinal fluid of patients (
52). A meta-analysis of PET studies using translocator protein binding, which is expressed during the inflammatory response, found significantly elevated tracer binding in patients with schizophrenia compared with control subjects, with a small to moderate effect size (Hedges’ g=0.31), but no difference in volume of distribution was detected (
53), although another study (
54) did not report evidence of neuroinflammation in the dorsolateral prefrontal cortex or hippocampus in unmedicated patients after controlling for relevant genetic polymorphisms. Two studies have reported evidence of neuroinflammation in psychosis spectrum patients with recent onset and patients with acute illness (
55,
56). Interestingly, these alterations are not found in medicated patients with chronic illness (
57,
58), suggesting a dynamic imbalance. This is further supported by a number of diffusion imaging studies reporting increased extracellular free water, a proxy of inflammation, which appears more prominent in the early illness compared with chronic stages (
59–
62). Taken together, biomarkers related to immune dysregulation may be dynamic, in that they capture pathological processes that are present only during illness onset or psychosis exacerbations.
Human connectome studies have provided data concordant with the hypothesis that brain network dysconnection is fundamental to psychosis (
63–
65). The dysconnectivity model proposes that NMDAR-mediated disturbances in the excitation/inhibition balance lead to altered functional network architecture, which in turn results in the symptoms of schizophrenia (
66,
67). A number of studies have demonstrated a relationship between brain network dysconnectivity and the development of psychotic symptoms (
62), symptom severity (
68), and response to antipsychotic pharmacotherapy (
69–
71). Lending additional support to this model are studies that report disruption in functional brain networks following experimentally induced NMDAR hypofunction in healthy human subjects (
35,
72,
73). While dysconnectivity classically is explained by changes in neurotransmitter systems and aberrant modulation of synaptic efficiency (
66), contemporary theories also consider the underlying anatomical connections. Prominent neurodevelopmental models postulate that genetic and environmental factors may affect early white matter developmental trajectories followed by the onset of psychosis, which then results in further reductions in white matter integrity (
74–
77). Those perturbations result in white matter development disruptions and altered behavioral phenotypes, supporting this neurodevelopmental model (
78). For example, mice lacking the chemokine receptor fractalkine exhibit a transient reduction of microglia and a deficit in synaptic pruning, which results in decreased connectivity between the frontal cortex and hippocampus and deficits in social interactions (
79). Importantly, diffusion imaging studies report associations between white matter abnormalities and disease severity across symptom dimensions (
80–
84), poor response to antipsychotic treatment (
85), and worse overall outcomes (
86,
87).
Similarly, cortical gray matter volume loss, which may be most prominent in fronto-temporal regions, is consistently found as a hallmark feature that is already present at illness onset and may become progressively worse with longer illness duration (
88,
89). Abnormalities in brain maturational processes mediated by environmental factors are hypothesized to underlie gray matter alterations (
90). Subchronic NMDAR antagonism (
91,
92) and maternal immune activation models (
93) have resulted in gray matter reductions in preclinical studies. Gray matter loss has been linked to disease severity across symptom dimensions (
94–
96) and overall poor clinical outcomes (
97), suggesting that it may be a viable candidate for diagnostic and prognostic biomarker development.
We have discussed a number of mechanistically plausible biomarker development targets, but it needs to be acknowledged that this is not an exhaustive list. Other targets are also plausible, such as oxidative stress or GABA dysfunction, where a disruption in the fast-spiking GABAergic interneurons has strong evidentiary support in the postmortem schizophrenia literature, and parvalbumin-positive interneuron dysfunction has been identified as a common factor behind several of the relevant animal models (
98,
99). As we next discuss, the specific utility of any of these markers may depend on the phase of illness and the specific clinical question to be answered.
Biomarkers for Risk Prediction
Correct identification of individuals at risk for psychosis in the prodromal phase provides a major opportunity for early intervention and disease prevention. However, when solely relying on clinically high-risk criteria, correct disease prediction is estimated to be 15%−30% (
100). In a two-center study examining structural MRI for predicting transition risk in patients at high risk for developing schizophrenia, Koutsouleris and colleagues (
101) demonstrated a statistically significant improvement in prognostic certainty over clinical assessments alone. The anatomical features associated with a later transition to schizophrenia included gray matter reductions in the prefrontal, cingulate, striatal, and cerebellar cortices. However, the study’s pooled sample was unusual in the fact that approximately 45% of the enrolled subjects transitioned to a psychotic illness, which is substantially higher than in a typical sample for this type of study (
101); replication in independent samples will therefore be important.
In a small study, Schobel and colleagues (
102) found that cerebral blood volume in the CA1 subfield of the hippocampus predicted clinical progression with a positive predictive value of 71% and a negative predictive value of 82%. The same group later reported that left anterior CA1 cerebral blood volume also predicted time to psychosis onset in high-risk patients, and demonstrated this to be a more sensitive marker of clinical outcomes compared with subthreshold psychotic symptoms (
46).
Thalamic glutamate measurements with MRS in a relatively small sample of ultra-high-risk individuals predicted the clinical course with an odds ratio of 0.52, such that higher baseline glutamate levels were associated with subsequent remission of prodromal symptoms (
103). In contrast, high-risk subjects who later developed a frank psychotic episode showed higher glutamate levels in the dorsal caudate compared with the nontransition group and control subjects. That study reported a surprisingly large effect size (Cohen’s d) of 1.39 (
104). Another small study showed that clinical high-risk individuals who later became psychotic had higher hippocampal glutamate levels compared with those who did not transition; the effect size (Hedges’ g) was 0.57 (
105), suggesting that brain regions are an important consideration when developing spectroscopy-based biomarkers.
Target Engagement Biomarkers
Target engagement biomarkers are meant to confirm desired therapeutic properties or to determine individualized dosing for a specific treatment. To qualify as a target engagement biomarker, it must measure a direct interaction between the treatment and the intended molecular or functional target in the central nervous system, which for medications is typically accomplished with PET. However, any neuroimaging outcome measure is theoretically an engageable target, as long as it has the potential to change measurably with a targeted intervention.
PET studies of the dopaminergic system provided the classic demonstration of target engagement for antipsychotic medications through several lines of converging evidence. These included establishment of abnormal dopaminergic function in patients with schizophrenia (
17,
108), discovery of a link between dopamine dysregulation and psychosis severity (
14), and demonstration of a reduction in dopamine stimulation of D
2 receptors with antipsychotic medication treatment (
109).
In the absence of a specific PET ligand targeting the glutamatergic system, a number of MRI markers are being used as a proxy for glutamate. The Fast-Fail Trials in Psychotic Spectrum Disorders biomarker project, based on the hypothesis of glutamatergic neurotransmitter system disturbances as a core pathological feature in schizophrenia (
18), approached validation of target engagement in two phases. In the first stage, three candidate biomarkers that are putatively sensitive to glutamatergic alterations were evaluated for their power to detect changes resulting from experimentally induced NMDAR hypofunction using a ketamine challenge (
110). The effect size was most robust and consistent across sites for a resting-state blood-oxygen-level-dependent (BOLD) signal response in the dorsal anterior cingulate cortex, suggesting possible utility of this measure as a biomarker for glutamatergic change. Importantly, a mean signal change of 0.5% fully separated subjects receiving ketamine from those receiving placebo. In the second stage, this marker was used to enrich the sample by eliminating subjects who did not demonstrate a ketamine-induced BOLD signal change before randomizing healthy volunteers to receive either pomaglumetad, a partial mGluR-2/3 agonist, or placebo. Here, the goal was to test the hypothesis that the drug would blunt the ketamine-induced BOLD response, indicating its glutamate target engagement, and to determine dosing of the medication. This stage of the study has now completed data collection; pending results will inform the decision for pomaglumetad to be advanced into further trials.
Biomarkers to Predict Response to Treatment or to Aid in Therapeutic Monitoring
This type of biomarker is intended to characterize patients in the context of a given treatment before it is started, and it may be used for patient stratification into biomarker positive and negative groups. In addition, these biomarkers may also be used to monitor the effectiveness of treatment or to predict deleterious side effect occurrence and probability of relapse (
111).
Antipsychotic medications principally act on dopamine D
2 receptors, which informed the dopamine hyperactivity hypothesis of schizophrenia. A number of studies reported a relationship between dopamine D
2 receptor occupancy at baseline and subsequent clinical response to antipsychotic medications (
15,
17), and a relationship between D
2 receptor occupancy with antipsychotic medication treatment and the extent of clinical improvement (
15,
16). Interestingly, the level of haloperidol-induced D
2 receptor blockage has also been found to be associated with the degree of extrapyramidal symptom severity and prolactin elevation (
15). Taken together, the data suggest that dopamine D
2 receptor occupancy is both a mediator and a moderator of antipsychotic treatment response.
The schizophrenia literature links greater reductions in gray matter volume to worse long-term clinical outcomes. In first-episode psychosis patients, a machine learning classifier based on gray matter morphometry found baseline gray matter volumes in the left and right parahippocampal gyri to be most important for predicting remission status after 6 months of antipsychotic treatment, with an accuracy of 79% (
112). Similarly, a multivariate morphometry study found that lower gray matter volume in the left and right inferior frontal gyrus and anterior insula predicted a lack of improvement in symptoms over 1 year (
113). The gyrification index, a gray matter morphometric feature acting as a surrogate of neurodevelopmental trajectories, was found to be altered in schizophrenia (
114) and possibly sensitive to antipsychotic exposure (
115). In first-episode psychosis, reduced gyrification in frontal, insular, and temporal cortices has been associated with subsequent poor response to antipsychotic treatment (
116), suggesting that it may be a useful predictor of treatment response.
A growing body of evidence suggests the potential to predict treatment response using glutamatergic neurometabolites measured with MRS. OPTiMiSE, a European multicenter study, found that higher baseline glutamate levels in the anterior cingulate cortex in first-episode psychosis patients were associated with greater symptom severity and lower likelihood of remission after 4 weeks of antipsychotic treatment (
117). This is consistent with a previous report by the same group finding 11% higher glutamate levels in the same region in a small group of patients with medication-resistant illness compared with a group with medication-responsive illness (Cohen’s d=0.76), although accurate classification of response status was not possible with this measure (
118). In a 2017 review of longitudinal spectroscopy studies, Egerton and colleagues (
119) reported an overall mean reduction in brain Glx (glutamate + glutamine) of 6.5% after antipsychotic treatment and suggested that antipsychotic treatment response may be associated with lower glutamate levels before treatment and greater reductions in glutamate with treatment. However, it is possible that effects are region specific, as others have reported that in the hippocampus, higher as opposed to lower baseline Glx may be associated with favorable treatment response (
25).
A promising resting-state-fMRI-based putative biomarker was recently described in a series of experiments examining functional connectivity of the striatum, a principal site of antipsychotic drug action. First, Sarpal and colleagues (
120) demonstrated an increase in striatal connectivity after 16 weeks of antipsychotic treatment, and this change was correlated with the degree of clinical symptom improvement. After this, the authors created a baseline “striatal connectivity index” that predicted subsequent treatment response in first-episode psychosis patients. This index was then tested for its utility in separating good from poor treatment responders in an independent cohort of hospitalized patients with chronic schizophrenia. Notably, this index showed a significant separation between good and poor responders, with a positive predictive value of 76% and a negative predictive value of 79% (
71). Extending this work, others later reported lower striatal connectivity in schizophrenia patients with antipsychotic treatment-resistant illness compared with patients with nonrefractory illness (
121).
A number of resting-state fMRI studies have suggested that the connectivity and topology of several other known brain networks (
69,
70,
122–
125) may also have utility in predicting response to antipsychotic treatment, although the performance of the majority of these putative markers remains to be established at the single-subject level. The only study to date investigating the accuracy of resting-state cortical connectivity at the single-subject level reported that baseline functional connectivity between the superior temporal cortex and other cortical regions predicted clinical response after 10 weeks of antipsychotic treatment, with a balanced accuracy of 82% (
126). Another group used a non-connectivity-based resting-state index, amplitude of low-frequency fluctuations (ALFF), to build a machine learning classifier predicting treatment response in patients with recent-onset schizophrenia. The authors reported that ALFF in the left postcentral gyrus/inferior parietal lobule predicted response status with an accuracy of 72.7%. A major strength of the study was the independent replication sample (using a different scanner) that showed a similar accuracy of 75% in predicting remission status (
127).
Challenges in Biomarker Development
Three challenges have limited the development of neuroimaging biomarkers in schizophrenia: internal heterogeneity, analytic approaches, and clinical utility.
First, one of the principal challenges of diagnostic biomarker research is that the standard nosology in schizophrenia is based on symptoms alone. There is an inherent tension between the effort to develop biomarkers for classic DSM-based diagnoses and the growing sense that it may be more fruitful to develop biomarkers that identify more biologically homogeneous subgroups of patients (
128). In an effort to overcome these inherent limitations, the Research Domain Criteria paradigm was introduced as an alternative framework for the investigation of psychiatric disorders (
129), in which disorders are considered in terms of disruptions of normal-range operation systems. Alternatively, the Bipolar-Schizophrenia Network on Intermediate Phenotypes initiative has incorporated a dimensional approach in an effort to analyze biomarker outcomes for disease definition and neuropathology in psychosis spectrum disorders (
130). Findings support the idea that neuroimaging-based biomarkers do not obey the boundaries set by symptom-based nosology, underscoring the limitations of diagnostic biomarker development studies conducted within the context of traditional diagnostic boundaries.
Closely related is the complexity of the underlying pathophysiology. Because of the high degree of intricate associations between neurotransmitter systems, immune systems, functional brain network architecture, and brain structure, a biomarker may reflect the result of multiple modulatory inputs rather than the primary etiological factor (
131). Unfortunately, multivariate approaches capitalizing on the vast amount of multimodal neuroimaging data available to detect representative pathway biomarkers assessing key pathological features such as NMDAR hypofunction, inflammation, or neuronal plasticity have yet to be developed. It also remains unclear whether putative biomarkers assessed in a clinically stable state or in a more actively psychotic state will be most informative. Furthermore, the cross-sectional assessment of the candidate marker may not be the most relevant because the change of a given marker over time in an individual may be most predictive. Additionally, it is possible that certain relevant biological signatures, such as alterations in spine density or synaptic integrity (
132,
133), are simply below the resolution that can be captured by existing imaging methods.
The second challenge lies in defining standard best practices for analytics, both in preprocessing of the original data and in the development of prediction rules. To the first point, the majority of conventional imaging indices are affected by scanner-specific measurement errors, which can have a significant impact on cross-site reliability. Furthermore, there is a lack of consensus in the field on how to harmonize imaging sequences or mitigate measurement error in the postprocessing stage across sites, which greatly reduces biomarker accuracy and reliability in multisite studies. To the second point, many biomarker studies do not report their results in the most rigorous fashion. The ultimate goal of every biomarker is to predict an unseen future: conversion to first-episode psychosis, response to a specific medication, long-term prognosis, and so on. The most rigorous approach to assessing that prediction performance is replication of a biomarker on a new data set, collected by researchers independent of the original team. Since that type of replication generally takes years, a close alternative is cross-validation: reporting a prediction model’s performance on new data it has never seen before (as opposed to the data originally used to build the model) (
134,
135). Without cross-validation, biomarker studies are susceptible to the problem of “overfitting”—building models that work very well on one specific sample but do not generalize to the larger clinical population. Unfortunately, cross-validation remains rare in schizophrenia biomarker studies, as it does in psychiatric biomarker research more generally.
The third challenge lies in determining the clinical utility of a biomarker. The decision to use a biomarker in clinical practice should be based on an expectation that it will have a positive health impact; therefore, measuring biomarker performance in generic terms is not sufficient for confirmation of clinical utility (
136). Demonstration of clinical utility is typically a two-step process that first determines the accuracy of a biomarker and then shows that using the biomarker information in managing patients, given the benefits and risks associated with the assessment, improves outcomes in a clinically meaningful way (
137). Prospective, confirmatory multicenter studies or decision-modeling approaches could be used to demonstrate clinical utility (
136,
137), but these types of studies are clearly a missing element in the evaluation of neuroimaging biomarkers in the field.
Of course, these challenges are not just specific to neuroimaging biomarker development in schizophrenia; they largely apply to neuroimaging biomarker research in general. Similarly, the following section, where we discuss next-generation biomarker development studies and reflect on best practices, is also applicable to the broader field.
Next-Generation Biomarker Development Studies
Several lines of research are well positioned to contribute to the advancement of neuroimaging biomarker discovery (
Figure 3).
Many of the routinely assessed imaging markers are difficult to link directly to relevant pathological processes. For example, glutamate levels measured with spectroscopy do not equal the amount of glutamatergic neurotransmission, but rather reflect the amount of neuronal, glial, and synaptic glutamate present in a voxel. BOLD imaging does not measure neural activity directly. It is a signal that stems from a complex interaction between blood flow, blood volume, and oxygen consumption related to neural activity. The field has demonstrated a commitment to the long-term goal of creating the next generation of imaging tools that allow us to map the brain and relevant pathological signatures on a much more granular level (
138), which will likely accelerate biomarker discovery. Continuing development of innovative technologies that are biologically validated and capture relevant features of complex neural circuits will provide unique opportunities. Here, radiotracer development holds distinct promise, as novel pharmacotherapeutics will likely target modulation of neurotransmitter systems. Development of specific ligands that capture principal pathophysiological processes such as NMDAR hypofunction or immune dysregulation could result in significant advancement of diagnostic and target-engagement biomarker research. Another strategy that may have greater short-term feasibility is to capitalize on already existing imaging techniques and leverage advances in computational modeling to increase the specificity of measures. For this strategy to be successful, vital components include testing the biological accuracy by rigorous histological validation and confirming disease relevance in preclinical models.
It will be critical to design studies that follow sound methodology to minimize bias and maximize precision (
139). Unfortunately, this is not a trivial undertaking with neuroimaging data, as factors such as magnetic field strength, sequence acquisition parameters, scanner variability, and data analysis methods affect measurements. Additionally, head movement has a negative impact on data quality in virtually all neuroimaging modalities (
140). A number of these problems can be mitigated by implementation of standardized image acquisition protocols, use of imaging phantoms as external reference if feasible, rigorous data quality control protocols, retrospective sequence harmonization (
141), and standardized data processing pipelines and data analytics.
Future studies would benefit from including efforts to cross-validate biomarkers in independent samples not only in patients with comparable clinical characteristics but also with various clinical presentations (acutely psychotic, clinically stable), at early and late disease stages, and with differential antipsychotic medication exposure. This type of data would be invaluable in delineating the temporal evolution and scope of a candidate biomarker.
Because an aggregate approach, either as a combination of multiple imaging modalities or the addition of information from serological or genetic markers, could improve biomarker performance, an important next step beyond independent replication will be to embed a number of biomarkers in a predictive model and to test overall performance, discrimination, calibration, and clinical utility of the model (
142). An excellent example of the important considerations of predictive models, in this case clinical risk prediction models for conversion to psychosis (
142), could also serve as a road map for building analogous models for disease risk or therapeutic response that are neuroimaging biomarker based.
Conventional statistical approaches for three-dimensional neuroimaging data rely on mass-univariate analyses (
143). Machine learning, a field in artificial intelligence, is designed to detect patterns in data with multivariate statistics aimed at making predictions at the single-subject level. To date, the majority of machine learning studies have focused on correctly classifying the presence of a disease state. However, the practical value here is limited, as those patients are already “correctly classified” through clinical assessments. Studies geared toward building models that can accurately predict the risk for disease development, clinical course, or a patient’s likelihood of responding to a given treatment may be more clinically useful but have been less common (
144). There are also a number of technical limitations that constrain interpretability of machine learning studies in schizophrenia. Because overfitting of data is a major source of bias, it is important that sample sizes are adequate when developing a model and that the model is then cross-validated as noted above (
145). Another limitation of machine learning approaches is that many studies use a “black box” approach, reporting sensitivity, specificity, and accuracy of an imaging classifier without providing spatial maps outlining relevant features of classifiers. Even though these types of complementary analyses are computationally expensive, they would make models more interpretable and add face validity to them. This idea of adding “explainability” to artificial intelligence techniques is a major priority for federal funders whose investments often shape a field (
146). Computer science holds immense promise in providing solutions for these technical limitations in the next generation of biomarker discovery studies. Opportunities are in the optimization of informatics infrastructures or design of user-friendly software interfaces geared toward neuroscientists with limited training in command-line based coding, and in interdisciplinary collaborations guiding appropriate implementation of multivariate statistics.
The literature on best practices in how to objectively and effectively evaluate the clinical utility of a neuroimaging biomarker in schizophrenia is sparse. Defining biomarker performance standards based on established decision theory concepts has shown potential to inform clinically relevant levels of sensitivity and specificity in cancer research. We would argue against arbitrary targets in favor of calculating target levels for a given intended use of a biomarker, for example, screening versus confirmatory testing. These calculations consider variables such as prevalence of the disease, which affects the true positive and negative predictive values, and the cost-benefit ratio of such tests (
147). Additionally, performance of a biomarker in one context may not be relevant to the setting of interest (
148). Development of calculators for estimation of individualized minimally acceptable biomarker performance indicators and necessary sample size based on these considerations could be invaluable in the design of future biomarker studies.
Lastly, to accelerate biomarker development, it is important to address transparency and reproducibility of research findings. On a policy level, many funding agencies now mandate sharing of generated data with the scientific community. Unfortunately, the sharing process is quite laborious at this time. Refining electronic data capture systems to allow highly automated sharing and integration of data sharing and analysis tools will improve reproducibility of imaging findings (
149). It will also be important to develop reporting standards containing the minimum recommendations for biomarker development studies, similar to those for randomized clinical trials (
150) and systematic reviews (
151), which are widely recognized in the scientific community. These guidelines help improve the quality of reporting in scientific journals by outlining essential items necessary for a clear and transparent account of research methods and results. Another interesting idea that could address reproducibility issues for future studies is the creation of a preregistration database for neuroimaging biomarker studies with the original hypothesis and its justification, similar to what is done in clinical trials (
152).
Moving forward, nationwide or multinational initiatives supporting large-scale studies that are developing targeted biomarkers have real potential to individualize clinical care in this complex neuropsychiatric syndrome. In the field of Alzheimer’s disease, the Alzheimer’s Disease Neuroimaging Initiative (ADNI), a private-public partnership, was designed to develop biomarkers for the early detection and tracking of the disease. The investment of $218 million to date has enabled ADNI to discover biomarkers for use in clinical trial subject selection and as surrogate outcome measures, to develop standardized protocols for use across multiple centers, to create platforms allowing open data access, and to make advances in the understanding of the relationship between biomarkers and disease progression (
153). This initiative could serve as a road map for the design of biomarker development studies for schizophrenia spectrum disorders.