A recent meta-analysis on high-quality randomized controlled trials (RCTs) of antipsychotic augmentation and monotherapy concluded that the only beneficial outcome related to antipsychotic polypharmacy is reduction of negative symptoms, which was found for aripiprazole add-on treatment (
8). Few differences in adverse effects were observed, including less insomnia and more prolactin elevation with D
2 antagonist augmentation, and reduced prolactin levels and body weight with aripiprazole augmentation. A recent RCT on amisulpride-olanzapine combination observed a beneficial effect on total score on the Positive and Negative Syndrome Scale for combination treatment compared with monotherapy, with effect sizes ranging from 0.29 to 0.40 (
9). Weight increase was more common in patients receiving amisulpride plus olanzapine than in those receiving amisulpride plus placebo, but this was attributable not to combining two drugs but to olanzapine per se
, since a similar weight increase was observed in patients receiving olanzapine plus placebo. However, it is important to note that the outcome in these RCTs is short-term symptom reduction and tolerability, whereas most polypharmacy occurs during maintenance treatment. Because RCTs typically last 3–6 months, they cannot provide information on long-term safety issues, which are attributable to relatively slowly developing adverse effects, such as weight gain leading to diabetes and ischemic heart disease. Since schizophrenia is a lifelong illness, the most important issue—in addition to quality of life and level of functioning—is long-term well-being, indicated by relapses and physical health problems leading to hospitalization or death (
10). Investigating these issues in RCTs, especially when the comparative effectiveness of a large number of specific drug combinations is of interest, would require thousands of patient-years, which makes these studies practically impossible to conduct. This problem can be solved in observational studies by using large nationwide databases and long follow-up periods.
Methods
Patients diagnosed with schizophrenia in inpatient care were identified from the nationwide Hospital Discharge register for the period 1972–2014 in Finland who were alive on January 1, 1996 (N=61,889). Diagnoses of schizophrenia were derived as ICD-10 codes F20 and F25 (corresponding to ICD-9 and ICD-8 code 295.x). Data for the cohort were linked via personal identification numbers on all inpatient stays (dates and diagnoses since 1972, from the Hospital Discharge Register), dispensed medications (from the Prescription register, 1995–2017), and deaths (Causes of Death register, 1972–2017). The follow-up period for this study started on January 1, 1996, for those diagnosed before that date and at the date of first diagnosis for those diagnosed later, and ended on December 31, 2017, or at the patient’s death, whichever occurred first. Permissions for this study were granted by the pertinent institutional authorities at the National Institute for Health and Welfare of Finland, the Social Insurance Institution of Finland, and Statistics Finland. Because the study was registry based and no contact was made with patients, informed consent was not required.
The study outcomes were nonpsychiatric hospitalization (main diagnosis other than ICD-10 codes F00–F99) and hospitalization due to diseases of circulatory system, referred to here as cardiovascular hospitalization (codes I00–I99). In cases where multiple diagnoses were recorded for a hospital care period, the main diagnosis was used. Main diagnoses are based on the clinical judgment of the treating physician. Additionally, we assessed risk of psychiatric hospitalization (hospitalization with codes F20–F29 as main diagnosis). These outcomes may occur multiple times for the same person (meaning that they are recurrent events).
Antipsychotic dispensing data were modeled into drug use periods by calculating a sliding average of daily dose in defined daily doses (DDDs) as defined by the WHO with the PRE2DUP method (
21) (see Table S1 in the
online supplement). A sliding average of dosage is used for smoothing local changes of dosage, and this calculation is controlled by expert-defined parameters that restrict average dosage estimates to clinically relevant dosages (i.e., a lower than minimum dosage leads to termination of drug use period). After antipsychotic use episodes were formed, temporal dosage estimates were calculated at each dispensing date as the sum of the dispensed DDD of the two previous dispensings divided by the outpatient time of these dispensings, resulting in a dosage estimate in DDDs/day (
15). Antipsychotic episodes were subdivided into periods when a certain dosage was used continuously in the following DDD categories: <0.4, 0.4–<0.6, 0.6–<0.9, 0.9–<1.1, 1.1–<1.4, 1.4–<1.6, and ≥1.6. These categories were designed around the most frequent clinically relevant dosages (0.5, 1.0, and 1.5 DDDs/day) by allowing small variation in use and inaccuracy of modeling (i.e., the dosage category 0.9–<1.1 describes 1.0 DDDs/day use).
Antipsychotic use periods were divided into two mutually exclusive groups, namely, monotherapy periods, when only one specific antipsychotic was used, and polypharmacy periods, when two or more antipsychotics were used concomitantly. A person may contribute person-time to both monotherapy and polytherapy use, and to multiple or even all dosage categories during the follow-up (see Figure S1 in the online supplement). Protopathic bias occurs when the treatment is changed because of the first signs of the forthcoming outcome (which has not yet fully manifested), leading to reverse causality. In addition, there is a delay of several weeks before the full treatment effect of antipsychotics is reached, disfavoring recently started medications. Therefore, we conducted a sensitivity analyses by excluding the first 30 days for all treatment options (from each dosage category of monotherapy and polypharmacy, and from no use).
Statistical Analysis
The analyses were done in a within-individual design (where all comparisons are conducted within the same individual, that is, each individual acts as his or her own control) in a stratified Cox model, which was run by the SAS
phreg procedure, calculating hazard ratios with 95% confidence intervals and p values (
22). Time was reset to zero after each outcome event (separately for each outcome type). The impact of factors that do not change in time, such as genetics and initial severity of the illness, were eliminated by the design, and the analyses were adjusted for time-varying factors, which were time since cohort entry, temporal order of specific antipsychotics (1st, 2nd, and 3rd vs. ≥4th), and use of antidepressants, benzodiazepines and related drugs, and mood stabilizers (carbamazepine, valproate, lamotrigine, lithium). Only data from patients who had the relevant outcome event contributed to the estimation of adjusted hazard ratios.
Comparisons between dosage categories were conducted by 1) applying nonuse of antipsychotics as a reference, and 2) comparing specific dosage categories of polytherapy to the same dosage categories as monotherapy (only patients who had both of these during the follow-up contributed to these analyses).
Sensitivity analyses for the highest dosage category (≥1.6 DDDs/day) were conducted by having a specific two-drug combination as a reference and calculating the risks for monotherapies of those two specific drugs when used at the same dosage (≥1.6 DDDs/day). For example, when clozapine-aripiprazole ≥1.6 DDDs/day was the reference, monotherapy with clozapine ≥1.6 DDDs/day and monotherapy with aripiprazole ≥1.6 DDDs/day were compared with that. Secondary analyses were conducted by excluding the first 30 days from each exposure (after dosage category changes and also from nonuse), excluding the first 365 days from each exposure (only for high-dose use), and stratifying the study population by whether they had polytherapy ever versus never during the follow-up. The results are reported as adjusted hazard ratios with 95% confidence intervals. Because there were seven dosage categories, the threshold for statistical significance was set at a p value of 0.0071 (0.05/7) for primary outcomes (p<0.05 for secondary outcomes).
Results
The mean age at cohort entry was of 46.7 years (SD=16.0), and the mean interval since first inpatient diagnosis of schizophrenia was 8.8 years (SD=9.0) (
Table 1). The median follow-up duration was 14.8 years (IQR=7.4–22.0 years). During the follow-up in outpatient care, monotherapy was used 45.9% of person-time, polytherapy 33.8% of person-time, and antipsychotic nonuse 20.3% of person-time. The most commonly used antipsychotics varied between specific dosage categories such that most common specific antipsychotics in the lowest dosage categories (both in monotherapy and in polypharmacy) were risperidone and quetiapine, whereas clozapine monotherapy was common in the middle dosage categories, olanzapine monotherapy in the highest dosage category, and olanzapine-quetiapine combination in the highest dosage category, followed by clozapine-aripiprazole, which was the second most common two-drug combination in the two highest dosage categories (1.4–<1.6 and ≥1.6 DDDs/day). About 46.4% of patients had used high-dose (≥1.6 DDDs/day) monotherapy, and 52.6% had used high-dose polypharmacy.
The most commonly used dosage category in terms of number of users and person-time of use was ≥1.6 DDDs/day (
Table 2), with the exception that the number of users for monotherapy was highest for the 0.6–0.9 DDDs/day dosage. During the follow-up, 42,363 (68%) used polypharmacy, 48,832 (79%) used monotherapy, and 46,631 (75%) had nonuse of antipsychotics. Of those who used polypharmacy during the follow-up, 89% (N=37,632) also had monotherapy use, and of those who used monotherapy, 77% (N=37,632) also had polypharmacy use. Of those who used polypharmacy at the highest dosage category during the follow-up (N=32,556), 65% (N=21,255) had used the same dosage category as during monotherapy, and of those who used the highest dosage category of monotherapy (N=28,702), 74% (N=21,255) had used the same dosage category as during polypharmacy. For other dosage categories, the overlap varied by dosage group so that the lowest dosage groups showed the smallest overlap between poly- and monotherapies and the highest dosage showed the greatest overlap. The median duration of periods of monotherapy, polypharmacy, and nonuse was 135 days (IQR=35–505), 112 days (IQR=41–446), and 64 days (IQR=8–275), respectively. Among patients who had both monotherapy and polypharmacy during the follow-up, 51.5% had monotherapy before polypharmacy, and 48.5% had polypharmacy before monotherapy. The mean age at the start of the first monotherapy period was 45.8 years (SD=15.6), and the mean age at the start of the first polypharmacy period was 45.7 years (SD=14.9). The potential impact of the first outcome event on prescribing practices was assessed. The distribution of monotherapy and polypharmacy remained similar before and after the first nonpsychiatric hospitalization, as the proportion of person-time for monotherapy changed from 55% to 57%, and for polypharmacy, from 45% to 43%.
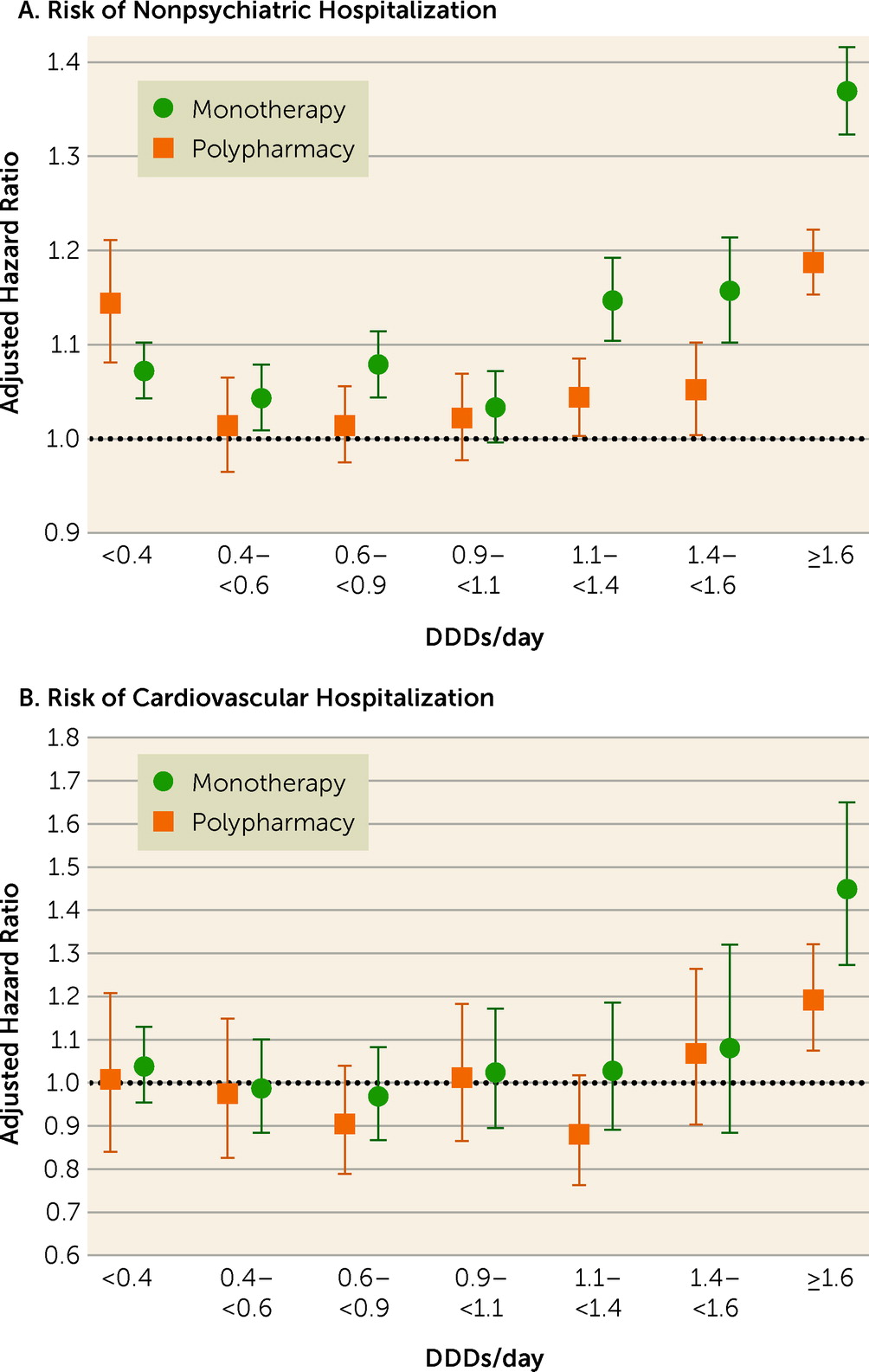
During the follow-up, 45,013 patients experienced nonpsychiatric hospitalization and 13,893 experienced cardiovascular hospitalization. Comparison of any polypharmacy use (all dosage ranges combined; median dosage, 1.54 DDDs/day [IQR=0.91–2.45]) with any monotherapy use (all dosage ranges combined; median dosage, 0.80 DDDs/day [IQR=0.39–1.33]) showed no statistically significant difference for nonpsychiatric (adjusted hazard ratio=0.99, 95% CI=0.97–1.00) or cardiovascular hospitalization (adjusted hazard ratio=0.98, 95% CI=0.92–1.05). Compared with nonuse of antipsychotics, high-dose (>1.6 DDDs/day) antipsychotic polypharmacy was associated with an increased risk of nonpsychiatric and cardiovascular hospitalization, but adjusted hazard ratios for high-dose monotherapy were even higher than for polypharmacy (
Figure 1,
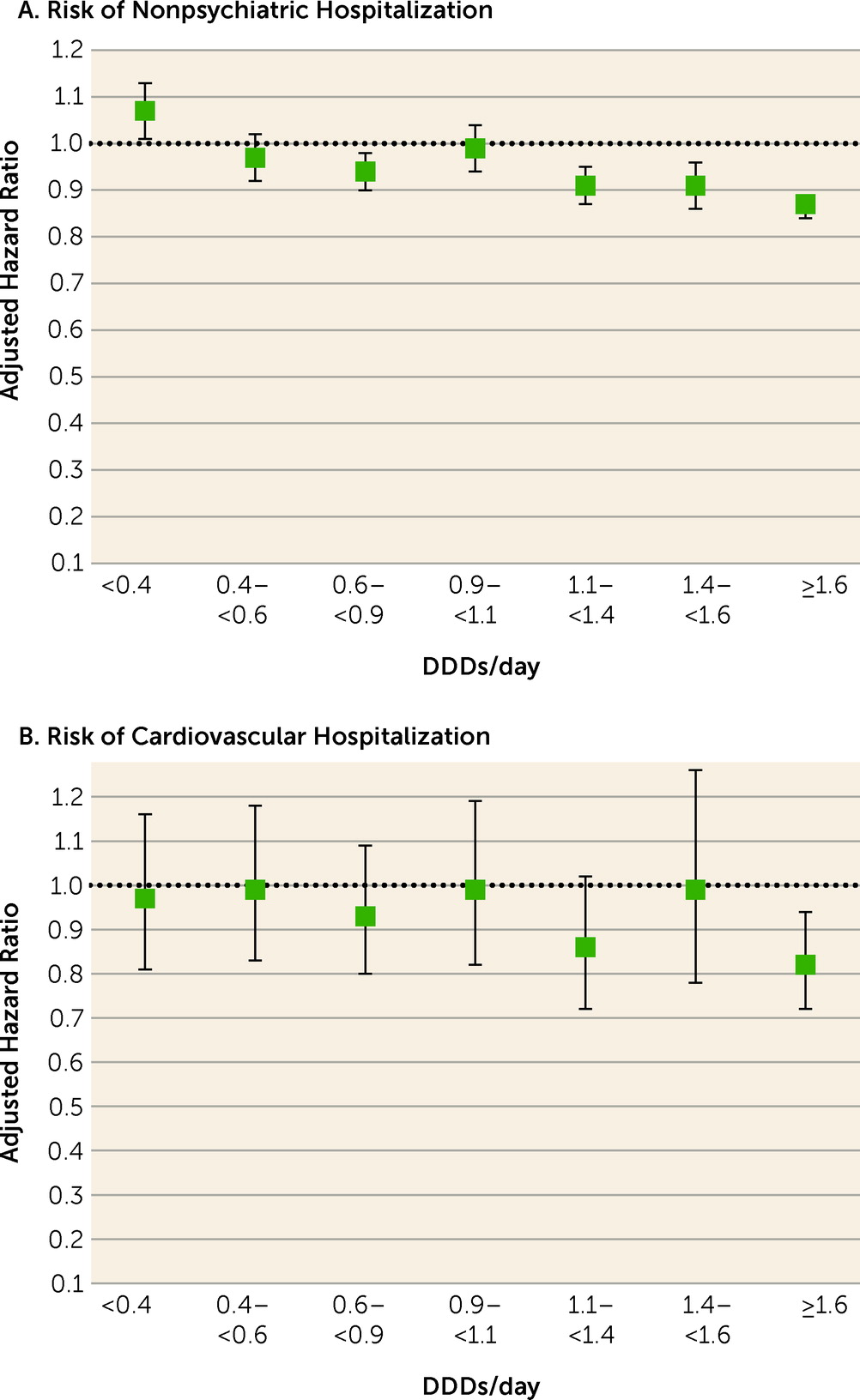
Table 2). Head-to-head comparison between specific polypharmacy categories with the corresponding dosage categories of monotherapy use showed that polypharmacy with dosages ≥1.1 DDDs/day was associated with lower risk of nonpsychiatric rehospitalization than the corresponding dosages of monotherapies (adjusted hazard ratios: for 1.1–<1.4 DDDs/day, 0.91, 95% CI=0.87–0.95; for 1.4–<1.6 DDDs/day, 0.91, 95% CI=0.86–0.96; and for ≥1.6 DDD/day, 0.87, 95% CI=0.84–0.89) (
Figure 2,
Table 3). In line with this, dosages ≥1.6 DDDs/day of polypharmacy use were associated with decreased risk of cardiovascular hospitalization compared with monotherapy at the ≥1.6 DDDs/day dosage category (adjusted hazard ratio=0.82, 95% CI=0.72–0.94). Monotherapy was not statistically significantly superior to corresponding polypharmacy in any comparison in any dosage category. However, monotherapy did have a lower, albeit not significantly lower, hazard ratio for nonpsychiatric hospitalization than polypharmacy at the lowest dosage range of <0.4 DDDs/day (for polypharmacy vs. monotherapy, adjusted hazard ratio=1.07, 95% CI=1.01, 1.13, p=0.02).
The results of sensitivity analyses comparing non-clozapine polypharmacy with non-clozapine monotherapy are presented in Table S2 in the
online supplement, showing adjusted hazard ratios similar to those in
Table 3. When the short (≤30 days) exposure periods of polypharmacy and monotherapy and periods of nonuse were omitted in the analysis, the adjusted hazard ratio for nonpsychiatric hospitalization during high-dose (≥1.6 DDDs/day) antipsychotic polypharmacy compared with monotherapy was 0.94 (95% CI=0.90–0.97, p=0.0007), and for cardiovascular hospitalization, it was 0.96 (95% CI=0.81–1.13, p=0.61) (see Table S3 in the
online supplement). When the first year was censored from the exposure (i.e., including only periods of use >1 year), the adjusted hazard ratio was 0.96 (95% CI=0.92–1.01) for the risk of nonpsychiatric hospitalization in the comparison between high-dose polypharmacy and monotherapy. Furthermore, when the analyses were stratified by whether the patient had polytherapy ever (N=42,042) versus never (N=19,487) during the follow-up, the adjusted hazard ratios associated with high-dose use were similar between these groups (see Table S4 in the
online supplement).
When two-drug combinations of polypharmacy were analyzed and compared with the same drugs as monotherapy in the highest dosage category, some combinations presented lower risk of hospitalization outcomes (see Table S5 in the online supplement). These were clozapine plus aripiprazole and olanzapine plus risperidone concerning nonpsychiatric hospitalization, and olanzapine plus quetiapine concerning cardiovascular hospitalization. Overall, long-acting injectables (LAIs) were used more often during high-dose polypharmacy than during high-dose monotherapy, as 38.5% of high-dose polypharmacy users had ongoing LAI use at some point during their high-dose polypharmacy periods, compared with 21.6% of high-dose monotherapy users. Similarly, benzodiazepines and related drugs (61.2% vs. 49.4%) and antidepressants (43.7% vs. 40.3%) were used more frequently by high-dose polytherapy than by high-dose monotherapy users. However, the figures for clozapine were the opposite, as 28.1% of high-dose polytherapy users had used clozapine, compared with 30.1% of high-dose monotherapy users.
Psychiatric hospitalization was analyzed as a secondary outcome (37,773 patients experienced this outcome during the follow-up at least once). Any antipsychotic polypharmacy was associated with a 6% lower risk of psychiatric hospitalization (adjusted hazard ratio=0.94, 95% CI=0.92–0.95). All dosage categories of both monotherapy and polypharmacy were associated with decreased risk of psychiatric hospitalization compared with nonuse of antipsychotics (see Table S6 in the online supplement). In head-to-head comparisons between antipsychotic polypharmacy and the corresponding dosage category in monotherapy, polypharmacy was associated with decreased risk of psychiatric hospitalization at both lower and higher dosages than standard dosage (0.9–<1.1 DDDs/day).
Discussion
To our knowledge, this is the first study to investigate the comparative safety of antipsychotic polypharmacy versus monotherapy in the treatment of schizophrenia in specific dosage categories. Our results showed that more than 40% of patients had used high-dose (≥1.6 DDDs/day) monotherapy, more than 50% of patients had used high-dose polypharmacy, and the vast majority of polypharmacy use time, in person-years, was at the highest total dosage category (≥1.6 DDDs/day). These figures underline the fact that antipsychotic polypharmacy is not confined to a small subgroup of patients but is a widely utilized practice in the treatment of schizophrenia. Our major findings were that when compared with monotherapy, the risk of nonpsychiatric hospitalization was lower for polypharmacy at total dosage categories above 1.1 DDDs/day, and that the risk of cardiovascular hospitalization was 18% lower for polypharmacy at the highest total dosage category when compared with monotherapy (corresponding to a 22% higher risk during monotherapy when compared with polypharmacy).
In other fields of medicine, polypharmacy is used commonly and is commonly recommended in treatment guidelines. For example, in the treatment of hypertension, combining two drugs with different mechanisms of action—such as an ACE inhibitor and a diuretic—leads to higher efficacy and better tolerability, and this practice is often preferred over using one medication at high or maximum dosage. In our study, nonpsychiatric hospitalization was considered a general safety measure. Concerning this outcome, the best combination in high-dose use was clozapine plus aripiprazole when compared with the corresponding monotherapies. This combination, composed of antipsychotics with different receptor profiles, aligns with the suggested basis of rational antipsychotic polypharmacy (
23). In terms of cardiovascular safety, the olanzapine-quetiapine combination performed best. It is plausible that this finding is attributable to lower olanzapine dosage during combination treatment than olanzapine monotherapy, resulting in fewer metabolic and cardiovascular adverse effects (
24). This practice is in line with a large meta-analysis of RCTs reporting that weight gain was 43% greater with olanzapine compared with quetiapine (
25) and another meta-analysis reporting that weight gain in olanzapine treatment did not plateau, even at high dosages (
26). It is logical to assume that adverse effects are milder when any two antipsychotics are used concomitantly at dosages of 1 DDD each compared with use of one antipsychotic at 2 DDDs/day. Although the total antipsychotic dosage is the same in both options, adverse effects are less likely to add up during polypharmacy because adverse effect profiles may differ between different antipsychotics. Since clozapine is associated with the highest burden of metabolic adverse effects and is used only in treatment-resistant illness, we conducted a secondary analysis comparing non-clozapine polypharmacy and non-clozapine monotherapy. The results of this analysis were well in line with the primary analysis including clozapine treatment, indicating that polypharmacy results are not driven by clozapine.
Our results are in line with all previous large studies showing better long-term outcomes for antipsychotic polypharmacy in general compared with monotherapy (
4,
12–
14). No previous study has compared polypharmacy with monotherapy in terms of dosage. In this study, we showed that polypharmacy is safe relative to monotherapy within the same dosage categories in terms of nonpsychiatric and cardiovascular hospitalizations and that polypharmacy is associated with better outcomes than monotherapy at total dosages ≥1.6 DDDs/day. Since the majority of polypharmacy was used at dosages ≥1.6 DDDs/day (e.g., corresponding to more than 8 mg risperidone or 16 mg olanzapine), it is probable that the reason for combination therapy has been to achieve higher efficacy and better tolerability than by increasing monotherapy dosages to the same high dosages. Because the greatest benefit in terms of lower risk of nonpsychiatric, cardiovascular, and psychiatric hospitalizations for polypharmacy compared with monotherapy was observed in this high-dose category, it is apparent that this approach is useful. Furthermore, these results call into question the safety of high-dose antipsychotic monotherapy use.
The results on standard dosage use showed no difference between polypharmacy and monotherapy in any of the outcomes studied. In the comparison between any polypharmacy (all dosage categories combined) and any monotherapy, no statistically significant difference was observed for the risk of nonpsychiatric or cardiovascular hospitalization. This was apparently attributable to substantially higher median dosages during polypharmacy (1.54 DDDs/day for polypharmacy vs. 0.80 DDDs/day for monotherapy). Our results showed that while monotherapy was used more in all dosage categories below 1.6 DDDs/day, polypharmacy was about twice as common as monotherapy in the highest dosage category, in terms of person-years of use. It is natural that when several antipsychotics are used, the total dosage is higher than during monotherapy.
Our results must be interpreted in the context of the study’s strengths and limitations. These results are based on a Finnish nationwide cohort that included all patients who had been hospitalized with a schizophrenia diagnosis. Therefore, the generalizability of the results is not an issue for Finland, and probably not for other higher-income countries with similar health care systems, but the results may not apply to middle- and low-income countries without full reimbursement of medication costs for patients with schizophrenia. In our cohort, the patients had their first hospitalization due to schizophrenia in their late 30s. Although the incidence of first hospitalization peaks before the age of 25 in Finland, the mean average age at the first hospitalization is more than 10 years later, explained by a relatively large proportion of late-onset illness among women (
27). Our analyses were based on antipsychotic prescriptions dispensed from pharmacies, and it was not possible to know how much of the dispensed medication had actually been used. However, blood level analyses have shown that our drug use modeling method determines actual medication use rather accurately (
28). Because observational studies are prone to selection bias, we used within-individual analysis to eliminate selection biases related to patient characteristics, such as sex, genetics, and initial severity of the illness. Although we adjusted for the duration of the illness, the temporal order of treatments, and concomitant use of antidepressants and benzodiazepines, it was not possible to exclude residual confounding in this nonrandomized study design. However, it should be noted that our knowledge of many severe adverse effects of pharmacological agents is based solely on observational studies. For example, no randomized trials have been conducted on the association between clozapine exposure and agranulocytosis. It should be noted that our results on polypharmacy are based on those patients who had used polypharmacy at some point during the follow-up. However, polypharmacy was used very often in this study population, and even the overlap between polypharmacy and monotherapy groups within the high-dosage category was large. Our results show the associations between medication exposure and the risk of any somatic and cardiovascular hospitalization, but it was not possible to determine how large a proportion of these hospitalizations were attributable solely to adverse effects of the medications. In addition, we could not rule out differences in physical health status as a potential source of bias.
One possible explanation for lower risk of cardiovascular hospitalization during polypharmacy is that the patients might be more adherent to medications used to reduce cardiovascular outcomes during polypharmacy than monotherapy, as has been observed previously during use versus nonuse of antipsychotic medication within the same person (
29). On the other hand, it is generally believed that patients requiring polypharmacy are more severely ill than those treated with monotherapy, which implies that this explanation may be unlikely. Given the vast complexity of measuring adherence to multiple medications and medication classes simultaneously over long periods, this issue was beyond the scope of this study. LAIs were used more often during high-dose polytherapy than during high-dose monotherapy, and in theory, lower plasma concentration peak levels during use of LAIs compared with during use of oral antipsychotics could contribute to better tolerability. However, a recent meta-analysis of 92 RCTs including 22,645 participants did not show any evidence of better tolerability for LAIs over oral antipsychotics (
30). While this might have resulted in better adherence and therefore a lower risk of psychiatric hospitalization, it should have resulted in a higher risk of cardiovascular and other nonpsychotic hospitalization caused by adverse effects of antipsychotics. Therefore, LAI use is not the only explanation for our findings on lower risk of somatic hospitalization during high-dose polypharmacy. However, it is possible that high-dose oral monotherapy may result in higher antipsychotic blood levels than high-dose polytherapy (including with LAIs), and that there is a shift in the dose-response relationship between the two forms of therapy, such that high-dose polytherapy is more comparable to middle-dose monotherapy.
Because many adverse effects related to antipsychotic use develop later during long-term treatment, the adverse outcome during a certain treatment period may be attributable partly to the preceding treatment period. Since patients are typically treated first with monotherapy and then later switched to polypharmacy if they do not respond well, this would disfavor polypharmacy. Also, because response to antipsychotic treatment decreases during the course of illness (
15), and because monotherapy is usually used as the first treatment line and polypharmacy later, the illness stage–related bias as well as protopathic bias disfavors polypharmacy. Therefore, we adjusted for time since cohort entry as well as temporal order of antipsychotic treatments in the statistical analysis. Polypharmacy may also represent an attempt to treat adverse effects caused by monotherapy; for example, when clozapine monotherapy has resulted in weight gain, aripiprazole can be added to prevent further weight gain and to allow a decrease in clozapine dosage. Therefore, the physical health problems induced during previous antipsychotic monotherapies may be classified as having been caused by the addition of the second antipsychotic, and the more beneficial outcome related to polypharmacy is underestimated. On the other hand, the opposite occurs as well: polypharmacy is switched to monotherapy because of health problems. Although it was not possible to know the reasons for switches, we adjusted the order of treatments in order to minimize bias.
Another source of bias is that in our analysis all overlapping antipsychotic use periods are labeled as antipsychotic polypharmacy, although some of the shorter overlapping periods are actually switches from one monotherapy to another monotherapy. It is possible that patients whose psychiatric symptoms responded well during cross-titration (related to intended switch) continued their treatment with two antipsychotics, leading to bias in the analysis. However, this applies only to psychiatric hospitalization, and not to safety issues indicated by nonpsychiatric and cardiovascular hospitalization, which were our primary outcome measures. In addition, the analyses omitting the first 30 days from all exposures showed results for high dosages similar to those of the main analyses. The result was also quite similar when the first year of exposure was omitted, but it was not statistically significant because of the lower number of events and wider confidence intervals. Our interpretation is that hospitalization outcomes in these analyses suffer not only from lack of statistical power (the original adjusted hazard ratios being within the confidence interval of that sensitivity analyses), but also because the risk estimates are diluted when the first month or the first year of exposure (when the acute adverse effects take place) is omitted. On the other hand, the lack of statistical power also applies to our finding of a nonsignificantly lower hazard ratio of monotherapy versus polypharmacy for nonpsychiatric hospitalization at the lowest dosage range. Analyses stratified by ever versus never use of polypharmacy during the follow-up and risk of safety outcomes showed similar significantly higher hazard ratios for monotherapy at the highest dosage compared with nonuse, and thus did not imply any inherent differences between patients exposed to polypharmacy and those with monotherapy or nonuse only.
Possibly the most important bias in our analysis is related to asymmetry concerning estimated end of use for polypharmacy versus monotherapy. Antipsychotics are typically dispensed to cover a 90-day supply, so on average, abrupt discontinuation due to safety issues will happen between 1 and 90 days after last dispensing (about 45 days on average), before the medication would run out if used as instructed and as observed in our drug use modeling, which assumes that all dispensed medications have been used. Therefore, if antipsychotic polypharmacy is switched to monotherapy before the full dispensed supply of the discontinued antipsychotic has been used, the following hospitalizations are allocated to polypharmacy—although they actually occurred during monotherapy. Likewise, if monotherapy is switched to polypharmacy, hospitalizations from the first day after the switch are allocated to polypharmacy, which may also lead to underestimation of the more positive outcome observed for polypharmacy. However, especially during longer time periods, adverse effects induced during previous polypharmacy may manifest after a switch to monotherapy.
We did not have information on the frequency of monitoring visits of the patients, and if patients with polypharmacy were more intensively monitored, that might contribute to the risk of hospitalization. Although more intensive monitoring could prevent some hospitalizations (e.g., measurement and treatment of hypertension), in some other cases more intensive monitoring could be associated with more nonpsychiatric hospitalizations, because conditions such as atrial fibrillation would be detected by doctors but not necessarily noticed by patients themselves. Our analysis included an assumption that the time-varying covariates are multiplicatively related to the hazard, and this can be considered a potential limitation of the study.