Conducting the largest RNA sequencing study of PTSD in the human brain to date, we identified downregulation of microglia-related transcripts and immune-related coexpression modules in both the cortex and amygdala. We identified notable reductions in specific transcripts encoding neuromodulators that are associated with GABAergic neuron function, but there was also evidence for increased expression of transcripts associated with both excitatory and inhibitory neurons. Collectively, the findings contribute evidence supporting the involvement of immune signaling, neuroinflammation, and inhibitory neuron function in MDD and PTSD.
Discussion
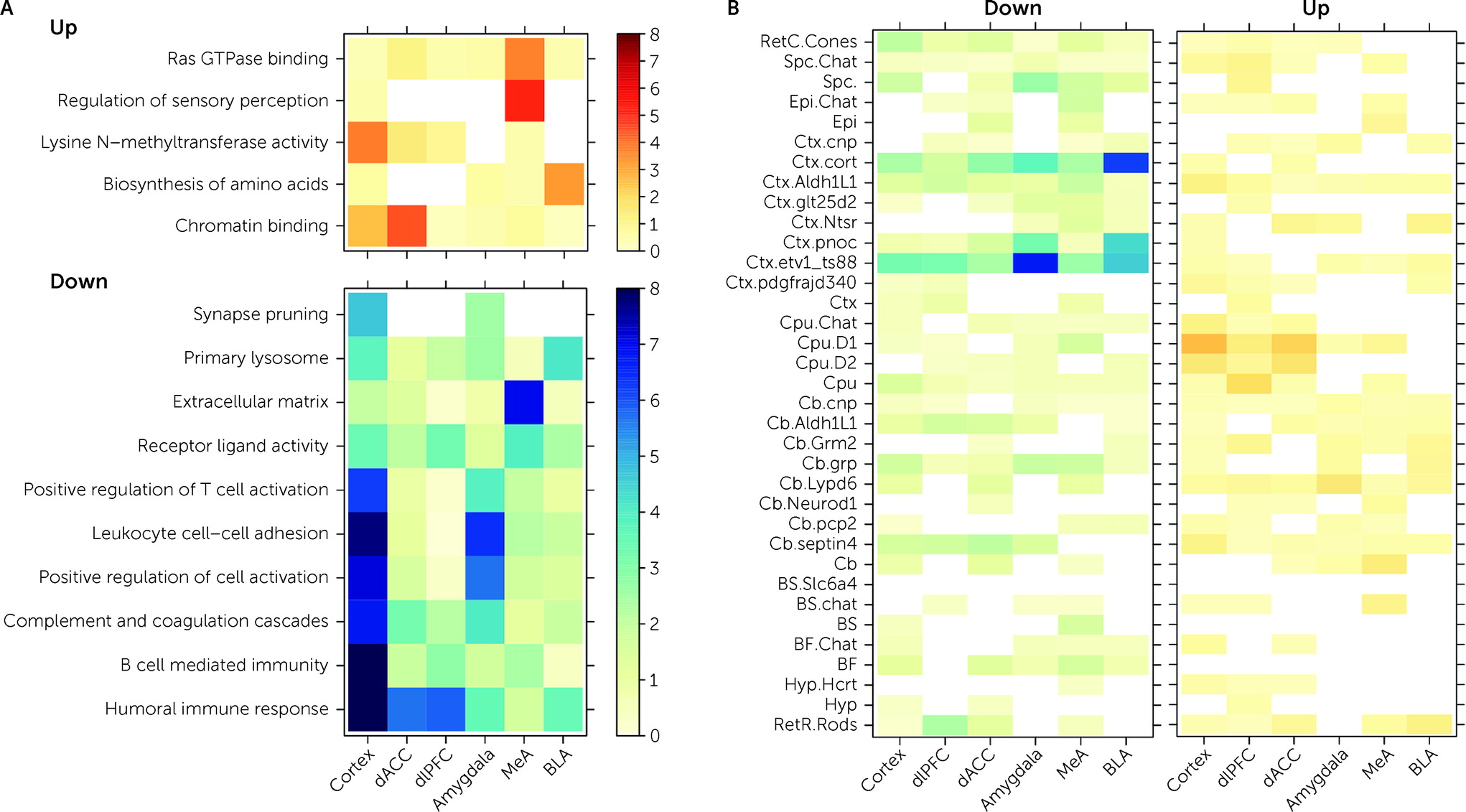
The goal of this study was to identify shared versus divergent in transcriptional patterns within and across PTSD and MDD in the prefrontal cortex and amygdala. We identified a limited number of DEGs specific to PTSD, with nearly all mapping to cortex versus amygdala. PTSD-specific DEGs were enriched in gene sets associated with immune-related pathways and microglia and with subpopulations of GABAergic inhibitory neurons. While we identified a greater number of DEGs associated with MDD, most overlapped with PTSD, and only a few were MDD specific. These findings provide supporting evidence for involvement of immune signaling and neuroinflammation in MDD and PTSD pathophysiology and extend evidence that GABAergic neurons have functional significance in PTSD.
Decreased expression of genes included in immune-related Gene Ontology sets were associated with PTSD diagnosis in both cortical and amygdala brain regions (
Figure 3A). CSEA using mouse cell-specific markers and snRNA-seq data from human brain demonstrated enrichment of these DEGs, with decreased expression in PTSD among microglia profiles (
41,
42). Genes with decreased expression in MDD donors compared with neurotypical control subjects showed analogous enrichment of immune-related processes using both gene set enrichment analysis and CSEA, and there were no enrichments for the immune and microglia-related genes when contrasting PTSD and MDD, suggesting that decreased expression of immune processes and microglia involvement are not specific to PTSD. The downward direction of dysregulation was somewhat surprising, considering that higher pre-trauma levels of C-reactive protein (a marker of blood inflammation) have been reported to predict elevated PTSD symptoms after trauma (
48). Furthermore, elevated levels of selected markers of low-grade blood inflammation have been reported in a meta-analysis of PTSD studies (
49). However, over time, and with repeated exposure to chronic stress and trauma, immune function may become dysregulated in a myriad of ways, with neuronal, glial, and peripheral systems attempting to compensate for immune activation and increased inflammation (
33,
50–
52). While decreased expression of the microglial immune transcriptome and/or reductions in microglial cell ratios due to chronic immune dysregulation are possible explanations for the present data, we noted that many of the genes included in the associated immune Gene Ontology sets encode proteins with known immunosuppressive activity. This could also explain the somewhat paradoxical finding of decreased expression of immune-related genes. For example, in the immune-related regulation of cell activity category, we identified 13 member PTSD DEGs (
IL1RL2,
DPP4,
IGFBP2,
TGFBR2,
TAC1,
MDK,
CD4,
PTPN6,
TESPA1,
IGF1,
ITGAM,
TYROBP, and
ITGB2), of which seven have potential immunosuppressive activity (
DPP4,
TGFBR2,
CD4,
IGF1,
ITGAM,
TYROBP, and
ITGB2) (
53). These observations do not support a high level of microglial immune activation in chronic PTSD or MDD in cortex or amygdala, but they do suggest dysregulation or possibly a compensatory response to stress.
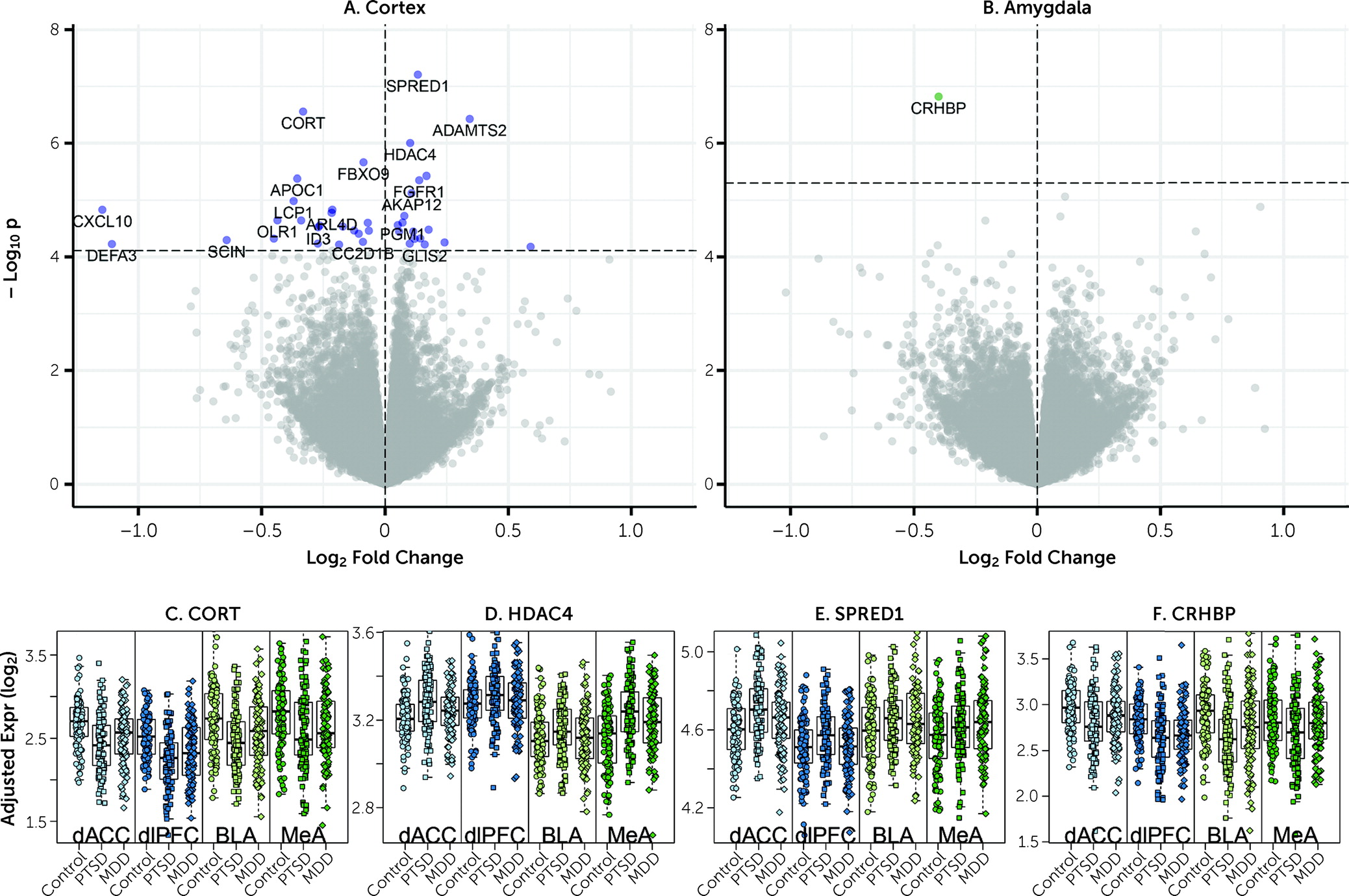
We observed downregulation of
CORT mRNA across all four subregions in individuals with PTSD.
CORT encodes the secreted neuropeptide cortistatin, which is expressed in the cerebral cortex, hippocampus, and amygdala in a subset of GABAergic neurons (
38,
54). Loss of cortistatin cells in mice causes spontaneous seizures, demonstrating that these cells provide strong inhibitory control (
55,
56). In the rodent, cells expressing cortistatin constitute a subset of SST-expressing neurons (
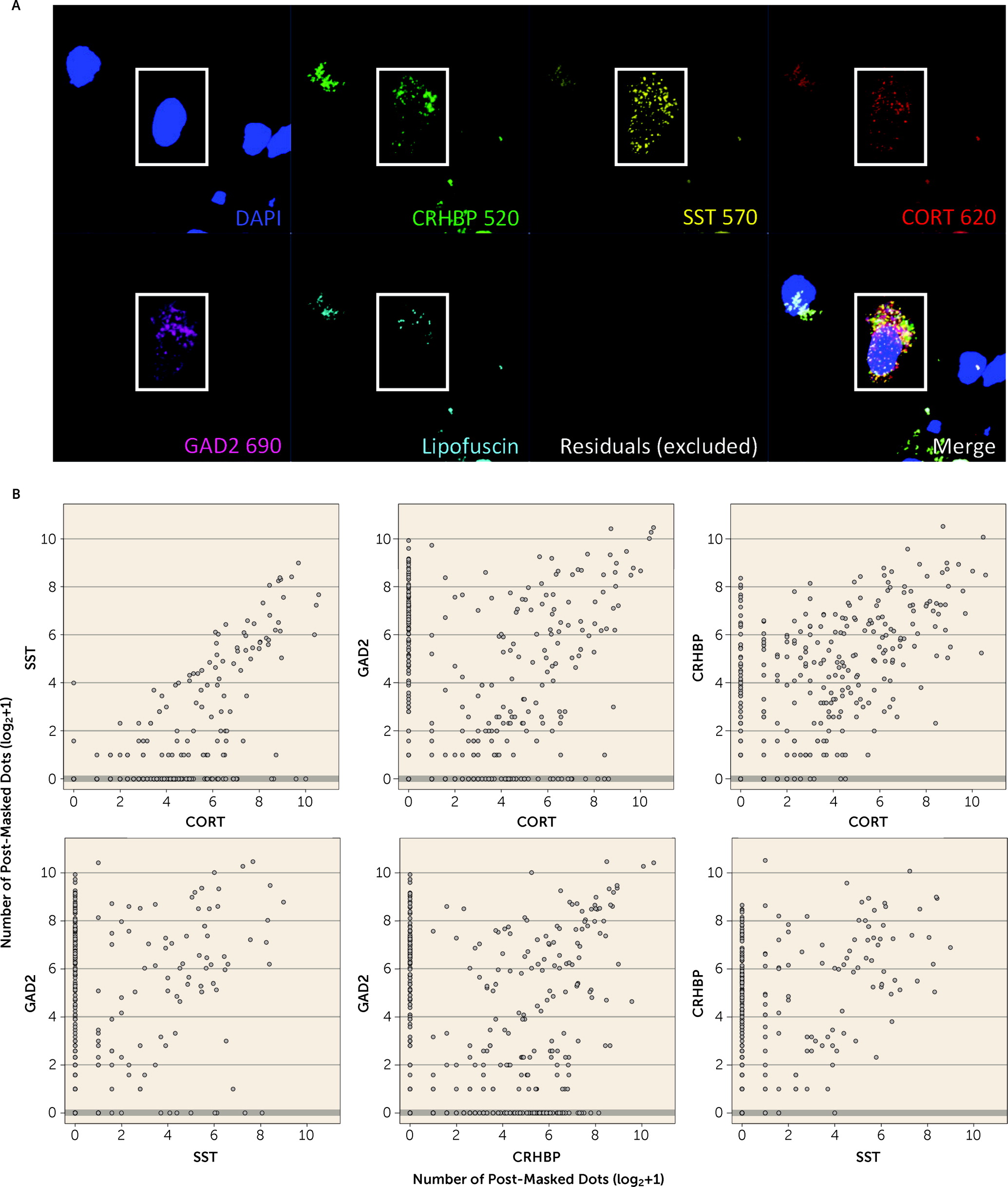
55), and in human brain we confirmed coexpression of
CORT with GABAergic inhibitory neuron markers (
GAD2,
SST, and
CRHBP). Decreased
CORT and
SST expression were previously reported in amygdala of female postmortem human brain donors with MDD (
57), and our CSEA analyses showed enrichment of genes differentially expressed in PTSD in cortistatin-expressing cells. We also identified enrichment of PTSD DEGs with specific inhibitory neuron clusters from snRNA-seq data in human amygdala that have been associated with anxiety and HPA axis function (
42,
58). WGCNA further implicated inhibitory neuron function, in line with the gene set enrichment results applied directly to PTSD DEGs. Decreased expression of
CORT,
SST, and
CRHBP mRNA provides additional support for the hypothesis that GABAergic neuron dysfunction is mechanistically associated with PTSD (
22). Strong evidence implicates GABAergic neurons in controlling fear-related behaviors in preclinical animal models relevant for PTSD and other trauma-related disorders by controlling neural activity and synaptic plasticity in cortico-amygdala circuits (
23). For example, firing of excitatory cells that project from the BLA to the frontal cortex is under tight regulation by local GABAergic inhibitory neurons (
25), which provides negative feedback regulation of the BLA to control both the expression and extinction of fear (
26,
59). Strong evidence links somatostatin signaling and
SST-positive cells in cortico-amygdala circuits with threat perception and fear memory processing (
60–
64).
CRHBP, which we showed to be coexpressed with
CORT and
SST in GABAergic neurons in the human brain, was the only gene consistently downregulated in PTSD versus neurotypical control subjects across both subregions of the amygdala.
CRHBP encodes corticotropin-releasing hormone binding protein, which sequesters and antagonizes CRH signaling (
39). The robust DEG signal for
CRHBP is interesting given many studies implicating the stress hormones corticotropin-releasing hormone (CRH) and cortisol in PTSD (
65–
68). CRH activates the release of adrenocorticotropic hormone (ACTH), which stimulates production of cortisol to control the body’s response to stress and trauma, which is important given that stress is a leading risk factor for the development of PTSD (
69). Additional genetic support for a critical role of the stress axis in PTSD comes from recent genome-wide association study associations of
CRHR1 (
70), which encodes the primary CRH receptor—CRF
1—and CRH binds to CRF
1 to mediate the behavioral and endocrine responses to stress exposure.
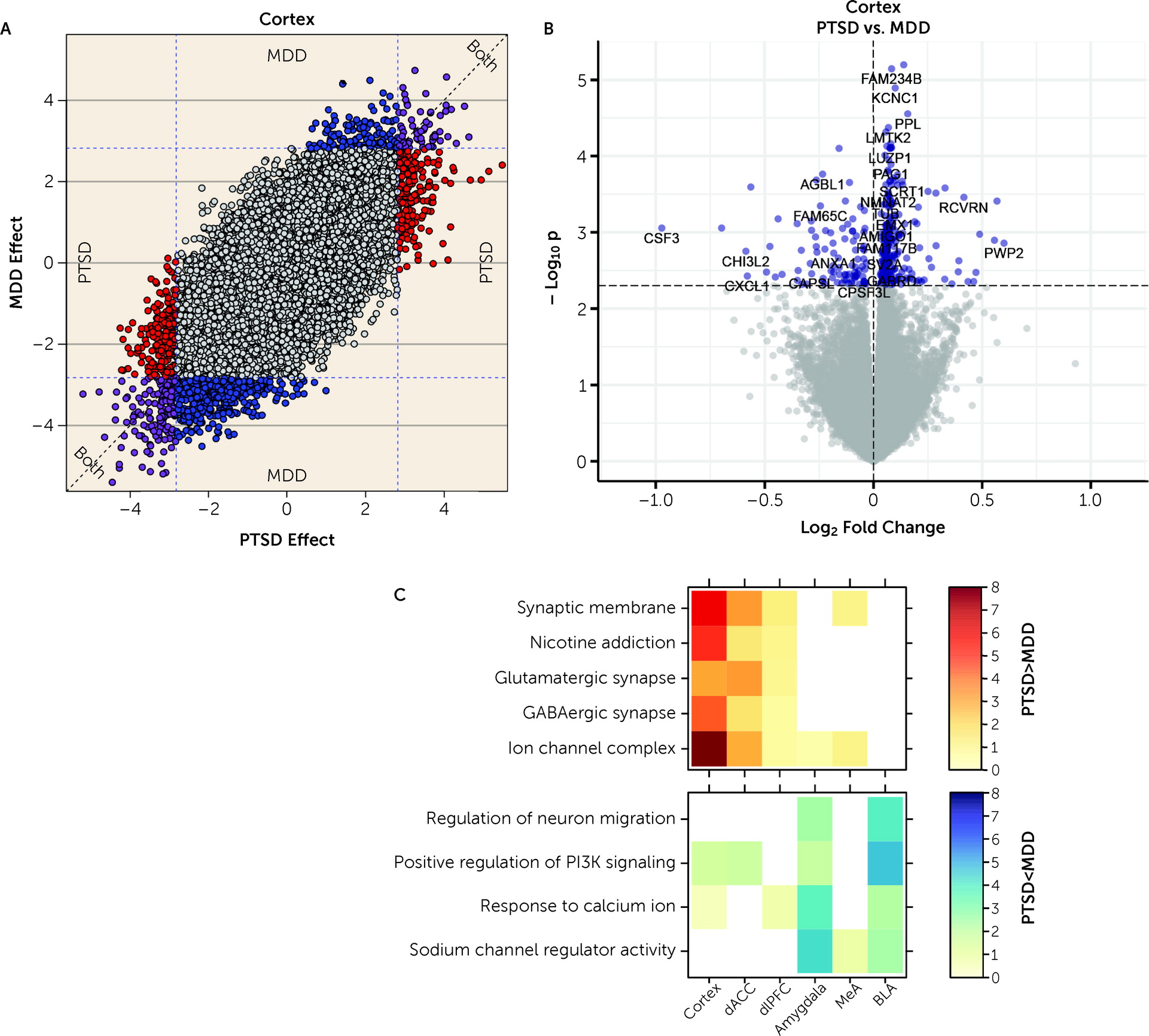
We further ran a number of analyses to better identify gene expression differences that were selectively associated with PTSD but not MDD. In general, we identified more DEGs for MDD than PTSD, particularly in the cortex (which were primarily driven by the dACC). However, gene expression differences were highly concordant between the two diagnoses, with most highly significant DEGs showing the same directionality of effects (i.e., log2 fold changes) in both diagnoses. Marginally selective between-diagnosis DEG gene sets included more highly expressed glutamatergic synapse-related DEGs in PTSD cortex (driven by the dACC) and more highly expressed neuronal activity-related DEGs in MDD amygdala (driven by the BLA). Differences between the two diagnoses were more prominent in WGCNA analyses, where seven potentially overlapping modules showed PTSD-specific enrichment (Cortex_ME31, dlPFC_ME20, dACC_ME9, Amygdala_ME2, MeA_ME3, BLA_ME2, BLA_ME13) and five modules showed MDD-specific enrichment (dlPFC_ME11, dACC_ME3, dACC_ME7, MeA_ME9, BLA_ME20).
DEGs from a recent RNA-seq study of human postmortem PTSD tissue by Girgenti et al. (
40), which used a partially overlapping set of donors (see below), provide support for top DEGs identified here. For example, within our combined cortical PTSD analyses (see
Figure 1), six of the seven most robustly affected transcripts comparing PTSD and control subjects (
CORT,
HDAC4,
CRHBP,
ADAMTS2,
FBXO9, and
APOC1) were directionally consistent and at least marginally significant in this previous data set. These genes further showed decreased expression in MDD versus control subjects in the present study, with at least marginal significance, suggesting that these particular findings may be related to shared pathophysiological changes accompanying PTSD and MDD. A key upregulated gene identified in the Girgenti et al. study,
ELK1, was significantly upregulated in both cortical regions in the present study, and
SST, identified as robustly downregulated in several regions of the cortex in the Girgenti et al. study, was in the top 10 of all downregulated transcripts in both dlPFC and dACC here.
ADAMTS2, the second highest upregulated DEG in the combined cortical sample, was the top upregulated gene in the dACC and the third most upregulated in the dlPFC in the Girgenti et al. study.
HDAC4, a top DEG, has been associated previously with both PTSD and rodent models of PTSD (
71,
72). The Girgenti et al. study also identified enrichment of downregulated PTSD-associated DEGs using CSEA that were related to GABAergic neurons and their molecular functions.
In addition to these common elements, the present results extend previous findings from Girgenti et al. (
40) in several key areas. First, this study extended the search for differential gene expression beyond the cortex and into the amygdala, a relatively understudied brain area in postmortem human brain research with high relevance to PTSD. Second, we provide compelling evidence implicating
decreased expression of immune-related genes and associated processes in PTSD and MDD compared with neurotypical control subjects. This is an important observation because it runs counter to most expectations for immune response directionality. We further refined these cellular enrichments in GABAergic neurons more specifically to
CORT-positive interneurons, which we subsequently validated with RNAscope. The cell type analyses in the present study provide direct evidence of these enrichments by interrogating DEGs directly against cell type-specific genes from both human and mouse studies, complementing the indirect strategy taken by Girgenti et al. of first identifying genes in discrete coexpressed modules and then associating those genes with both PTSD DEGs and cell type-specific genes separately (such that different genes captured the cell type vs. PTSD signal in the same module). Third, we believe our larger sample size (more than twice as large in all diagnostic groups), obtained from a single postmortem brain collection under identical sample ascertainment and inclusion criteria, refined several of the clinical associations identified by Girgenti et al. We identified more similarities than differences between PTSD and MDD and replicated this finding across both amygdala and cortical subregions, with far less sex-specific diagnosis-associated signal than discussed in the earlier study. Contributing to both the similarities and differences between the two studies was the fact that 77 donors were shared across the studies, although the two studies used different hemispheres, independent dissections and RNA extractions, and different data analysis pipelines. Over half (53.8%) of the donors in the Girgenti et al. study overlapped with one-quarter (23.7%) of the donors in the present study.
Therefore, it might seem counterintuitive that we identified many fewer DEGs in this much larger study, particularly with overlapping donors. We believe these differences can be accounted for by our more conservative statistical analyses, including the modeling of both diagnostic groups in a single statistical model against the neurotypical group, which further accounted for robust observed and latent confounders. The differential expression models in the Girgenti et al. study (
40) only adjusted for age, RNA integrity number, postmortem interval, and race, and did not account for sequencing-derived RNA quality metrics and other latent confounders, which can greatly increase false positive rates in human postmortem brain gene expression studies (
36). For example, this less comprehensive statistical model applied to our larger data set resulted in 1,243 DEGs in the dlPFC, 1,719 DEGs in the dACC, 1,813 DEGs in the BLA, and 10,283 DEGs in the MeA for PTSD at FDR<0.05, which is many more genes than obtained with our more conservative approach. There has been some debate regarding the optimal methods of latent variable correction in these types of postmortem studies, including the potential for “overcorrection” (
73). A major analytic element of the present investigation was the use of quality surrogate variable (qSV) analysis to identify and correct for expressed sequences that are particularly prone to degradation in human postmortem brain (
36). The qSVs utilized here were defined from the top 1,000 degradation-susceptible expressed regions generated from independent time course experiments. Dropping the qSVs from our main analyses resulted in 209 DEGs in the dlPFC, 43 DEGs in the dACC, 62 DEGs in the BLA, and 1,054 DEGs in the MeA (at FDR<0.05) for PTSD in the present data set. It is, however, possible that if sex or disease-associated interactions related to gene transcript degradation exist, use of qSVs may have limited the emergence of these genes as DEGs and contributed to the differences between the present findings and those discussed in the Girgenti et al. study (
40). Similarly, in MDD, the most prominent previous report of differential gene expression (
74) actually identified no DEGs when correcting for multiple testing via the FDR (from the supplementary tables included with that study), making it difficult to assess replication of our DEGs using previously published data sets. While these issues may seem rather nuanced, they nevertheless have important consequences for identifying DEGs in human postmortem RNA-seq data sets and require careful consideration in past and future work.
Limitations of this work include potential underrepresentation of female donors in the neurotypical control group (21.1% female) compared with the case group (∼50% female). While we adjusted for sex in differential expression analyses, secondary analyses did suggest some potential differences in diagnosis effects across sex. Our cohort included donors with only a PTSD diagnosis or only an MDD diagnosis, as well as PTSD donors with a comorbid MDD or bipolar disorder diagnosis. However, we did not have subjects with only a bipolar disorder diagnosis. Furthermore, as is common in most psychiatric postmortem human studies, psychotropic medications, substance use, smoking, and suicide were more common in the MDD and PTSD groups, and further work will be needed to investigate their potential influence on brain gene expression patterns.
In summary, these analyses of the largest postmortem brain cohort of patients with PTSD and MDD to date highlight microglia and other immune cell types as having potential functional significance in PTSD, and provide additional evidence for dysregulated neuroinflammation and neuroimmune signaling in MDD and PTSD pathophysiology.