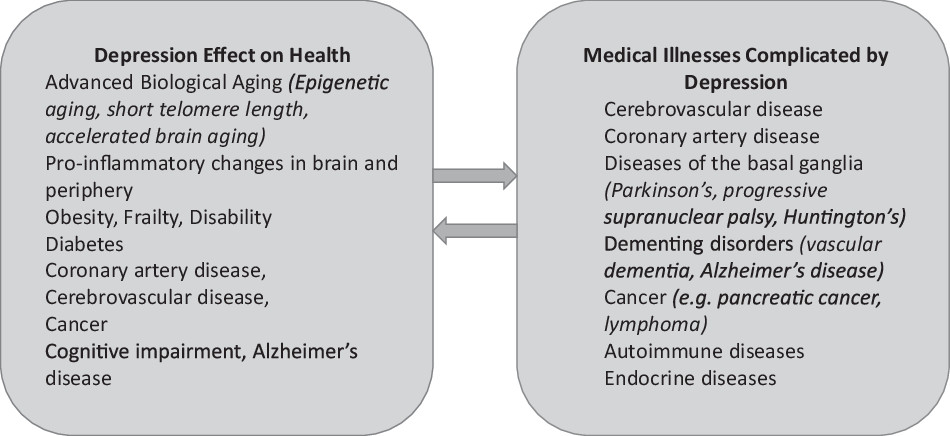
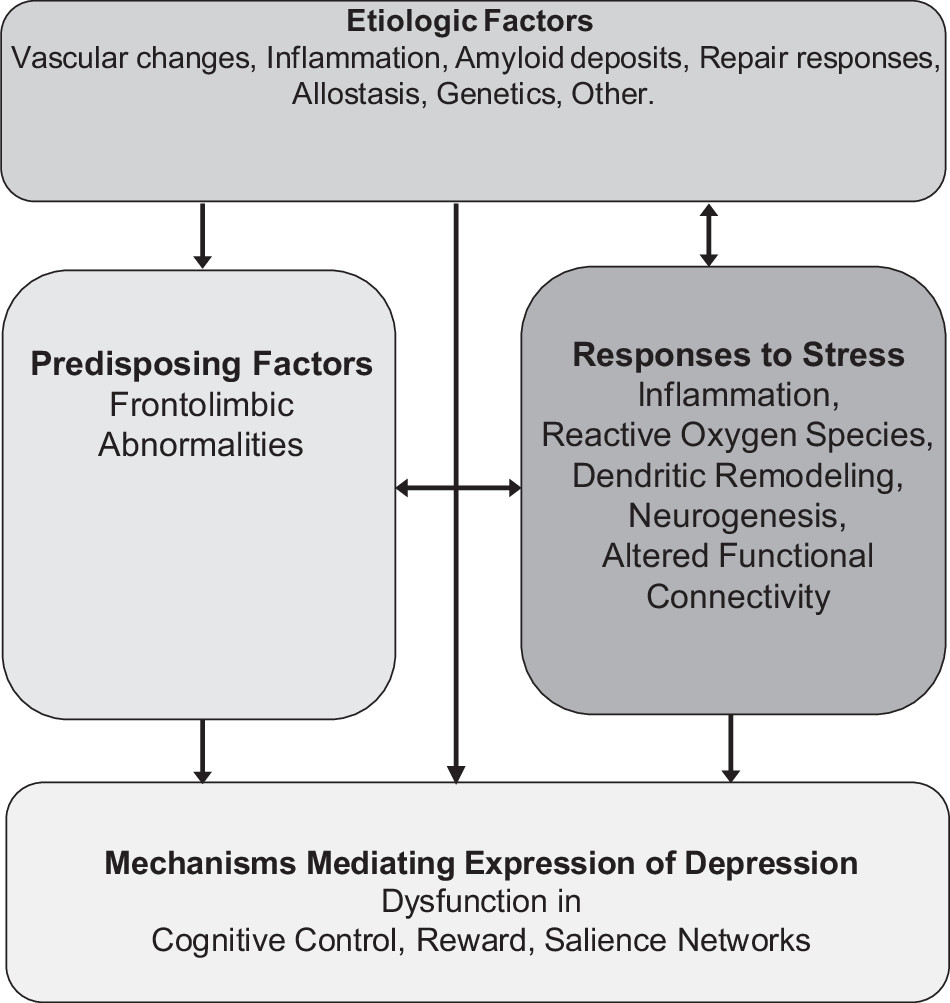
A working model of LLD postulates that the depressive syndrome represents the clinical expression of dysfunction in reward, salience and cognitive control networks (
8–
11) (
Fig. 2). The degree of dysfunction in these networks may determine the intensity of symptoms related to mood, cognition, and/or motoric behavior and account for the heterogeneous clinical presentations of the late-life depressive syndrome. Abnormalities in overlapping and/or distinct networks within the frontolimbic system may serve as predisposing factors, facilitating the functional abnormalities mediating the expression of depression, and promoting chronicity and relapse (
12). Genetic factors, aging, and disease-related processes (e.g., inflammation, vascular disease, amyloid accumulation)(
7,
13–
15) may serve as etiological factors by either directly promoting dysfunction in reward, salience, and cognitive control networks and/or by compromising frontolimbic networks predisposing to depression. Many of the etiological factors start in mid-life, e.g., hypertension, diabetes, obesity, vascular and hormonal changes, amyloid deposition, inflammatory responses, changes in neuroplasticity and synaptogenesis. Late-life and mid-life is often associated with medical and psychosocial problems at the individual (pain, unemployment, elder mistreatment, divorce/widowhood, poverty, social isolation) but also at the community level (rising costs/fixed income, limited access to health care, crime). These stressors may lead to inflammatory responses, increased reactive oxygen species, suppressed neurogenesis, and promote apical dendritic atrophy in the medial prefrontal cortex, and altered functional connectivity (FC)(
16). Stress responses may, then, lead to depression directly by triggering dysfunction in the reward, salience, and cognitive control networks, by promoting frontolimbic abnormalities pre-disposing to depression, or by increasing aging or disease related processes serving as etiological factors of depression directly (e.g., through allostasis (
17)) or indirectly through neglect of health. This model has served to organize testable hypotheses of relationships among etiological, predisposing, and stress related factors and mechanisms mediating the behavioral expressions of LLD and course of illness.
The syndromes described below are based on hypotheses related to the working model of LLD. Although described separately for simplicity, some of their mechanisms overlap and additional mechanisms may even be at play. For example, the depression-executive dysfunction syndrome is the clinical expression of frontostriatal dysfunction often contributed by cerebrovascular dysfunction, abnormal inflammatory responses, and perhaps amyloid deposition. By the same token, the vascular depression syndrome may present with symptoms originating from frontostriatal dysfunction caused by vascular lesions disconnecting networks related to mood regulation and executive functions, as well as reduced cerebral blood flow and inflammatory responses. Data-driven multidimensional approaches began to identify mediators of cognitive impairment of patients with LLD. Machine learning of proteomic data and measures of structural brain abnormalities and brain amyloid-β (Aβ) PET scans showed that cognitive impairment in LLD is related to greater cerebrovascular disease along with abnormalities in immune-inflammatory control, cell survival, intracellular signaling, protein and lipid homeostasis, and clotting processes (
18). Finally, a senescence associated phenotype consisting of 22 proteins was found elevated in LLD and associated with medical and cognitive burden (
19) indicating another source of vulnerability.
The depression-executive dysfunction syndrome hypothesis
A depression executive dysfunction (DED) syndrome has been described in older adults with distinct clinical presentation and poor response to antidepressants (
20). Approximately 30% of depressed older adults have abnormal performance in tests of verbal fluency, response inhibition, novel problem solving, cognitive flexibility, working memory, and/or ideomotor planning (
11,
21) (
Table 1). The depression profile of DED is characterized by anhedonia, psychomotor retardation, pronounced disability, lack of insight, and suspiciousness but less prominent depressive ideation and a mild vegetative syndrome (
22–
24). This presentation is consistent with disruption of frontal-subcortical networks. Depression frequently develops in disorders of subcortical structures, including vascular dementia, Parkinson’s disease, Huntington’s disease, supranuclear palsy and basal ganglia calcification, and stroke of the caudate head. White matter hyperintensities (WMH) are common in geriatric depression and often located in subcortical structures and their frontal projections (
25). Diffusion tensor imaging studies of LLD identified microstructural abnormalities in white matter tracts that connect the prefrontal cortex with subcortical and posterior cortical regions, which have been linked to executive dysfunction (
26,
27). Low metabolic activity and resting FC have been observed in the dorsal anterior cingulate cortex (dACC) and the dorsolateral prefrontal cortex (DLPFC) during depressive episodes in older adults (
8,
28). Tasks challenging the cognitive control network resulted in hypoactivation of the DLPFC in LLD and diminished FC between the DLPFC and the dACC (
28). Hypoactivation of the DLPFC resolved after SSRI treatment but decreased task-based FC persisted (
28).
Apathy is common in LLD and associated with executive dysfunction, disability, and poor antidepressant response (
29). The impairment in executive functions tests of DED patients may in part be due to the motivational disturbance of apathy. Alternatively, apathetic DED, may be a subtype of DED in which a shared neurobiological dysfunction leads to depressed mood, apathy, and executive dysfunction. Apathy is associated with reduction in white matter integrity in the anterior cingulum, fornix, and uncinate fasciculus (
30). Older apathetic patients with major depression had lower resting FC of the nucleus accumbens with the amygdala, caudate, putamen, globus pallidus, and thalamus and increased FC with the dorsomedial prefrontal cortex, the superior frontal cortex, and the insula than non-apathetic patients (
31). Further, apathetic depressed patients had lower resting FC of the dACC with dorsolateral and ventrolateral prefrontal cortices and higher FC with the insula and the orbitofrontal cortex than non-apathetic depressed patients (
31). Also, depressed elderly patients had decreased intrinsic resting FC of the salience network and an altered pattern of salience network FC to the right DLPFC node of the cognitive control network when compared to elderly non-apathetic depressed and to normal, elderly subjects (
9). These observations suggest that abnormal FC of the reward, salience and cognitive control networks underlies apathy of LLD.
Executive dysfunction predicts poor response of LLD to antidepressants and early relapse and recurrence (
32–
38). Subcortical WMHs are common in LLD and have been associated with both executive dysfunction and non-remission of LLD (
39). Diffusion tensor imaging showed that lower white matter integrity in distributed networks tracts (dorsal and rostral ACC, DLPFC, hippocampus, posterior cingulate, insula, neostriatum, and the midbrain, as well as select temporal and parietal regions) was associated with poor response of late-life major depression to a serotonin reuptake inhibitor (
26). Low resting FC within the networks supporting executive functions, but not within the default mode network (DMN), predicted persistence of depressive symptoms and signs, apathy, and dysexecutive behavior after treatment with escitalopram (
31). Similarly, lower activation in the DLPFC and other brain regions during in-scanner performance of the Wisconsin Card Sorting Test of executive functions predicted less favorable response to cognitive behavioral therapy in depressed, older adults (
40). The poor response of DED to antidepressants and the evolving understanding of its pathogenesis may guide the development of targeted interventions.
The vascular depression hypothesis
The ‘vascular depression’ hypothesis postulates that cerebrovascular disease may predispose, precipitate, or perpetuate some geriatric depressive syndromes (
13,
41). This hypothesis was based on the presence of cerebrovascular risk factors in many patients with LLD, the comorbidity of LLD with cerebrovascular lesions, and the frequent development of depression after stroke.
A clinical definition regards cerebrovascular risk factors or cerebrovascular disease as one of the cardinal features of vascular depression (
Table 2). Cerebrovascular risk factors are associated with WMH in healthy young adults (
42). Elevated systolic blood pressure has been associated with brain infarcts, gross infarcts, and micro-infarcts (
43). Vascular risk factors lead to vascular wall hypertrophy, increased intima media thickness, reduced arterial distensibility, and endothelial cell dysfunction (
44). Such vascular changes have been associated with poor response to antidepressants (
45). MRI stigmata of cerebral small vessel disease, (i.e., WMHs, lacunes, microbleeds, perivascular spaces, and cerebral atrophy) are associated with depression and incident stroke (
46). The Cardiovascular Health Study showed that persistence of depressive symptoms was associated with small basal ganglia lesions and large cerebral cortical white-matter lesions while worsening of depression severity was associated with subcortical white-matter lesions (
47). Greater arterial stiffness (carotid-femoral pulse wave velocity) was associated with depressive symptoms; this relationship was partly accounted by white WMH volume and subcortical infarcts (
48). Markers of progression of cerebral small vessel disease (WMH volume, subcortical infarcts, cerebral microbleeds, Virchow-Robin spaces, and total brain volume) over time were associated with new depressive symptoms in community elders (
49). Carotid plaque presence was associated with higher severity of depressive symptoms at a 10-year follow-up in men (
50).
A second cardinal feature of the clinical definition of vascular depression is either onset in late-life or worsening of the course of early-onset depression after the onset of vascular disease. Early onset does not preclude the diagnosis of vascular depression since history of depression increases the risk of vascular disease and stroke (
41,
51) and may promote inflammation (
52,
53) or epigenetic changes of genes related to vascular integrity (
14,
54,
55), suggesting that depression has a bidirectional relationship with vascular diseases.
Neurological signs and/or neuropsychological findings, usually executive dysfunction, are found in most patients with vascular depression depending on the location and extent of lesions. Late onset and absence of family history of mood disorders are expected in most cases but family history of mood disorders does not preclude vascular depression, since family history of mood disorders was shown to predispose to post-stroke depression (
56). Patients with vascular depression often present with retardation, anhedonia, lack of insight into their illness, and disability and are less likely to report feelings of guilt (
23,
57).
An MRI based definition of vascular depression requires presence of hyperintensities in the subcortical gray matter, deep white matter, or periventricular areas (
41,
57). Compromised white matter integrity is associated with LLD and predicts future depressive symptoms (
58). Depression has been associated with greater WMH severity in white matter tracts of the cingulum, uncinate fasciculus, and superior longitudinal fasciculus (
59,
60), as well as the frontal (
25) and temporal lobes (
61). Diffusion tensor imaging studies have shown reduced anisotropy in the DLPFC and the uncinate fasciculus of patients with LLD consistent with disruption of frontal and frontal-to-limbic white matter tracts (
62). Depressed older adults were shown to have decreased resting FC in the subgenual anterior cingulate cortex and increased connectivity in the dorsomedial prefrontal cortex and the orbitofrontal cortex;(
63) abnormal FC was correlated with greater WMH volume. High WMH burden in LLD was associated with greater activation of the subgenual cingulate in response to a facial expression affective-reactivity task, suggesting that white matter ischemic changes lead to limbic hyperactivation (
64).
Some neuropathology studies failed to identify a relationship between vascular brain lesions and depression. Neither lacunes nor microvascular ischemic lesions were related to occurrence of late-onset depression (
65,
66). Further, gross or microscopic infarcts were not associated with severity of depressive symptoms or change of depressive symptoms overtime (
67,
68).
WMH burden is associated with executive dysfunction and reduced activation of brain regions related to executive and psychomotor functions. Executive dysfunction was associated with bilateral WMH in the inferior frontal white matter, temporal-occipital periventricular white matter, and the anterior limb of the internal capsule, as well as scattered clusters in the prefrontal white matter (
69). WMHs were correlated with impairments in goal maintenance during a continuous performance task, but also with reduced activity in DLPFC and reduced connectivity of the DLPFC with task-relevant brain regions including middle frontal gyrus, and supramarginal gyrus (
70). In addition, WMHs were associated with increased activity in the anterior cingulate on a facial expression affective-reactivity task (
64).
A confluence of interacting events may lead to vascular depression. WMHs and microstructural abnormalities may damage fiber tracts including the cingulum, uncinate fasciculus, anterior thalamic radiation, and superior longitudinal fasciculus (
59,
60,
71,
72) and lead to disconnection and dysfunction of networks supporting affective and cognitive functions (
73). LLD patients were shown to have an anterior-posterior gradient in cerebral blood flow (CBF), with lower CBF throughout the frontal lobe but higher CBF in the parietal lobe, temporal lobe, thalamus, and hippocampus (
74). A similar anterior to posterior gradient was observed in the cingulate cortex. Aging related vascular pathology reduces blood flow velocities and decreases vasomotor reactivity (
75), compromising CBF. Large impairment in perfusion and autoregulation may result in WMH and gray matter lesions (
76). Circulating markers of endothelial dysfunction and flow mediated dilatation were correlated with depressive symptoms in a community of elderly population (
77). Pulsed arterial spin labeling showed that relative to healthy controls, remitted late-onset depressed patients had decreased cerebral blood flow in the precuneus and cuneus bilaterally and in the right fronto-cingulate-striatal areas, temporal, occipital, and parietal lobes, but increased CBF in the left frontal and temporal cortices and the cingulate gyrus (
78). A meta-analysis reported that higher levels of the plasma endothelial biomarker soluble intercellular adhesion molecule-1, WMH, cerebral microbleeds, and cerebral micro-infarctions are associated with depression; WMH were associated with incident depression (
79). A recent study reported an association of WMH with tumor necrosis factors alpha (TNF-α) and interferon gamma (INFγ) and macrophage inflammatory protein-1α (
80). Older adults with high homocysteine plasma level had increased risk of depression (
81). Aging related disruption of immune functions contribute to WMH burden and predispose to LLD (
82). Increased hypothalamic-pituitary-adrenal (HPA) axis function during depressed states may influence inflammatory responses. Amyloid deposition in and around cerebral blood vessels may change the integrity of blood–brain barrier and release of inflammatory mediators, which may damage the basal lamina and increase the risk of microhemorrhages (
7). In a transgenic mouse model of Alzheimer’s disease, cortical arterioles had Aβ accumulation, high tortuosity, and narrow caliber and their function was compromised (
83). Inhibition of Aβ oligomerization and fibrillization prevented both structural and functional impairment of the cortical microvasculature. Interactions among the above processes, and processes yet to be identified, may provide targets for prevention or treatment of vascular depression.
The inflammation hypothesis
The inflammation hypothesis posits that age-related and comorbid disease-related immune deregulation contribute to the etiology of LLD (
84) (
Table 3). Aging leads to a pro-inflammatory changes mediated by increased immune responses in the periphery, disruption of the periphery-brain immune communication, and an increased and discordant brain response (
85). Disruption in the periphery-brain immune communication, produces a disproportionate brain inflammatory response to peripheral immune stimulation, promoting a chronic proinflammatory state with increased activated and primed microglia, continuous production of proinflammatory cytokines IL-1β, IL-6, and TNF-α, and decreases in anti-inflammatory molecules (
86). Persistent activation of microglia leads to inefficient clearance of neurotoxic molecules, neuron loss, and reduction of neurogenesis (
87).
Cytokines induce indoleamine 2,3-dioxygenase, an enzyme that reduces serotonin production (
88). They also dysregulate the glutamate system, promote excitotoxicity and decrease production of neurotrophic factors, neuro-plasticity, and neurogenesis. In major depression, plasma C-reactive protein was correlated with concentrations of glutamate in the left basal ganglia (
89). Administration of the pro-inflammatory interferon alpha (IFN-α) increased glutamate in the basal ganglia of non-depressed subjects with hepatitis C;(
90,
91) changes in glutamate concentrations were in turn associated with anhedonia and psychomotor slowing. Cytokines contribute to oxidative stress, which damages glial cells in the prefrontal cortex and the amygdala (
92). Inflammation may cause resistance to glucocorticoids in immunocytes and their cellular targets (
93), disrupt glucocorticoid receptor function and increase inflammatory responses that further fuel depressive symptoms.
Inflammatory changes in the brain have been associated with depression. A PET study used F-FEPPA ligand to measure translocator protein total distribution volume (TSPO V
T), a marker of microglial activation (
94). Duration of untreated major depressive disorder was a strong predictor of TSPO V
T, as were total illness duration, and duration of antidepressant exposure. The combination of these predictors accounted for about 50% of variance in TSPO V
T in the prefrontal cortex, anterior cingulate cortex, and insula (
94). Increased cell adhesion molecule expression has been found in the DLPFC in LLD, an inflammatory response associated with ischemia (
95).
Inflammatory responses to immune challenge influence the function of emotional networks. A SNP encoding IL-1β has been associated with both reduced activity of the anterior cingulate and the amygdala in response to emotional probes and with poor response of major depression to antidepressants (
96). Patients treated with the cytokine INF-α exhibited greater dorsal anterior cingulate activation than controls;(
97) dysfunction of the anterior cingulate has been documented in geriatric depression (
98). Enhanced activation of the subgenual anterior cingulate cortex during emotional face processing and reduced FC of the subgenual anterior cingulate with the amygdala, medial prefrontal cortex and nucleus accumbens is modulated by IL-6 (
99).
Peripheral inflammatory markers are elevated in LLD and their levels are associated with severity of depression (
100) and with cognitive symptoms of depression (
101). A meta-analysis showed that peripheral levels of interleukin‐ 6 (IL‐6), tumor necrosis factor TNF-α, IL‐10, soluble IL‐2 receptor, C–C chemokine ligand 2, IL‐13, IL‐18, IL‐12, IL‐1 receptor antagonist, and soluble TNF receptor 2 were elevated and interferon‐gamma levels were lower in individuals with major depression compared to controls (
102). Elevated IL-6 is associated with increased suicide risk, with the highest levels of IL-6 correlating with the most violent suicide attempts (
103). High IL-1ra levels have been found in older adults with depressive symptoms and have been a risk factor for developing depressive symptoms during a 6 year follow-up (
104). Antidepressant treatment significantly decreased peripheral levels of IL-6, TNF-α, IL-10, and CCL-2(
105).
Pro-inflammatory changes have been documented in medical illnesses and health risk factors predisposing to LLD. Increased circulating inflammatory cytokines have been found in cardiovascular disease (
106). High body mass index and smoking have been associated with increased inflammatory markers in major depression (
102). Chronic stress, a precipitant of depression, exacerbates age-related increases in inflammatory responses and increases circulating IL-1β and IL-6 and cognitive impairment in elderly patients (
107). Although peripheral cytokines do not cross the blood–brain barrier, they send signals via molecular, cellular, and neural routes, which ultimately enhance brain inflammation (
15,
108). Aging may exacerbate the effects of stress in the brain, leading to behavioral and cognitive changes similar to those of depressive syndromes.
Is amyloid and Tau accumulation one of the mechanisms of LLD?
Several studies suggest that amyloid beta (Aβ) accumulation may predispose to LLD (
109). In cognitively unimpaired older adults, increased amyloid burden in the precuneus/posterior cingulate cortex were associated with depressive symptoms (
110). In community-dwelling, cognitively unimpaired elderly individuals, Aβ burden was associated with increasing anxious-depressive symptoms during a 1–5 year follow-up (mean = 3.8 years)(
111). Patients with a lifetime history of depression had amyloid accumulation in brain regions related to mood regulation (
112). Depression is associated with a high conversion rate of amnestic mild cognitive impairment (aMCI) to Alzheimer’s dementia (
113). Patients with aMCI and history of major depression had higher Aβ deposition, mainly in the frontal cortex, compared to patients with aMCI without history of major depression (
114). Alzheimer’s patients with history of depression had more amyloid plaques in the hippocampus than Alzheimer’s patients without depression (
115). Individuals with LLD had lower plasma Aβ
42 levels and a higher plasma Aβ
42/Aβ
40 ratio than did those without depression in the absence of cardiovascular disease and antidepressant use; high plasma Aβ
42/Aβ
40 increases the risk of Alzheimer’s disease (
116).
A single dose of citalopram decreased Aβ in the brain’s interstitial fluid in a dose-dependent manner in aged, transgenic (APP/PSI), plaque bearing, AD mice (
117). Chronic administration of citalopram arrested the growth of preexisting plaques and the development of new plaques by 78%(
117). In healthy individuals, acute administration of citalopram 60 mg slowed the production of Aβ in the CSF by 37% compared to placebo (
117). Community volunteers treated with antidepressants over a period of 5 years (mean: 34.5 months) had significantly lower amyloid load in brain PET scans than those who had never received antidepressants (
118). The length of antidepressant treatment prior to scanning correlated with lower plaque load. Finally, depression increases the risk of conversion of MCI to Alzheimer’s dementia (
113) and long-term treatment with antidepressants delays the conversion of mild cognitive impairment to Alzheimer’s dementia (
119).
Despite the above findings, several studies failed to identify a relationship between Alzheimer’s pathology and LLD. A neuroimaging study found no differences in cortical Aβ uptake or in the proportion of amyloid-positive subjects between depressed older patients and healthy controls (
120). Non-demented patients with prior depressive episodes had cortical Aβ levels indistinguishable from healthy controls (
121). An early neuropathology study reported no significant differences in plaque or tangle counts between subjects who were cognitively impaired and those who were unimpaired during their depressive illness (
122). A more recent study found no differences in neuritic pathology or neuronal density between the subjects with primary major depression and nondepressed comparison subjects (
123). Subjects with Alzheimer’s disease had fewer serotonergic neurons and more neuritic pathology, compared to depressed subjects and healthy controls but there were no differences between depressed and non-depressed Alzheimer’s disease subjects on these measures. Another neuropathology study found no significant association between depressive symptoms cognitive status, neuritic plaque, and neurofi-brillary tangle scores or their interactions (
124). Finally, there were no differences between LLD patients and healthy controls in CSF total and phosphorylated tau (
125). Discrepancies in the studies summarized above make it unclear whether and what aspects of neurobiological changes of Alzheimer’s disease are related to LLD.