Evaluating the Hypothesis That Schizophrenia Is an Inflammatory Disorder
Abstract
The investigation of immune system abnormalities in schizophrenia, although ongoing for decades, has become a popular area of research. The authors present a selected review of studies informing on schizophrenia as a potential inflammatory disorder, emphasizing replicated findings. The authors summarize evidence for inflammation over the illness course, discuss relationships between inflammation and psychopathology, present studies of imaging of neuroinflammation, consider inflammation as a marker of treatment response and treatment target, and review potential mechanisms for the effects of inflammation on the brain in schizophrenia. Although there is not clear and convincing evidence to support the assertion that schizophrenia is an inflammatory disorder, this area of study shows promise toward a greater understanding of the etiopathophysiology of this heterogeneous disorder.
Schizophrenia is commonly a chronic, debilitating disorder with lifelong consequences for affected individuals and families. Schizophrenia also is a heterogeneous disorder with respect to illness presentation, clinical course, and outcome. There has been renewed interest in investigation of inflammatory abnormalities in schizophrenia, stimulated in part by our increased understanding of brain-immune interactions in other chronic medical disorders. Recent genome-wide association studies have led to the identification of associations between genes involved in the regulation of the immune system and schizophrenia risk, perhaps most notably complement protein C4 (1–3). Prenatal maternal infection with bacterial, viral, and parasitic infectious agents is a replicated risk factor for schizophrenia (4), and the inflammatory response to infection is a potential common pathway underlying this association. There is a bidirectional association between autoimmune disorders and severe infections, which are associated with increased inflammation and schizophrenia (5, 6). There is evidence that patients with schizophrenia have inflammatory abnormalities in the blood, cerebrospinal fluid, and central nervous system (CNS), including levels of inflammatory markers, number of immune cell numbers, and antibody titers (7–10). Patients with schizophrenia may have an increased prevalence of certain comorbid infections and associated inflammation (11). Several trials have found that adjunctive treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) or other anti-inflammatory agents may be associated with improvements in psychopathology among some patients with schizophrenia (12, 13), and that baseline blood inflammatory markers may predict response to these agents (14, 15). Taken together, these findings support the plausibility that inflammation may be involved in the etiopathophysiology of schizophrenia, at least for some patients.
It is important to emphasize, however, that the evidence for inflammation in schizophrenia has been fraught with substantial heterogeneity, including negative studies and small magnitude of effect sizes for many associations. There are many potential explanations for such discrepancies, including small sample sizes of individual studies, different phases of illness studied, and potential confounding factors (e.g., smoking, obesity, medication effects, and medical comorbidities). Nevertheless, another important potential explanation is that inflammation may be present in only a subset of patients with schizophrenia. For example, many psychiatric hospitalizations in the late 19th and early 20th centuries were caused by psychotic symptoms in the context of advanced neurosyphilis. Subsequently, the recognition of an infectious cause of psychosis and the discovery and development of penicillin were game changers in the treatment of these patients. The identification and characterization of a potential subgroup of patients with schizophrenia and inflammation may thus hold promise of novel disease- and life-altering treatment approaches.
In that regard, two case reports provide compelling evidence for potential mechanistic associations between the immune system and schizophrenia. Sommer and colleagues (16) described a 67-year-old man with chronic lymphocytic leukemia and no past psychiatric history who developed new-onset psychosis status after receiving an allogenic peripheral blood stem cell transplant from a brother with chronic schizophrenia. Conversely, Miyaoka and colleagues (17) reported on a 24-year-old man who experienced prolonged remission of treatment-resistant psychosis—without any antipsychotic medication—after receiving a bone marrow transplant for acute myeloid leukemia. Of course, is it challenging to establish causality, but the bidirectional nature of these case reports provides support for the plausibility of such an association.
In this article, we present a selected review of studies informing on schizophrenia as a potential inflammatory disorder, with emphasis on replicated findings. Because a review of all aspects of immune function in schizophrenia is beyond the scope of the present work, the reader is also referred to related comprehensive reviews (18–22). We present a theoretical framework in an attempt to integrate these findings, and we suggest potential mechanisms whereby inflammation may play an etiopathophysiological role in a subset of patients with schizophrenia (Table 1).
| Category | Marker |
|---|---|
| Genetic risk factors | IL-1β, sIL-6R, CRP |
| First-degree relatives | sIL-2R, IL-6 |
| Prenatal maternal inflammation | IL-1, IL-6, IL-8, IL-10, CRP |
| Inflammation in childhood | IL-6 |
| Clinical high risk for psychosis | IL-1β, IL-6, IL-12, CRP |
| First-episode psychosis | IL-1β, IL-1RA, sIL-2R, IL-6, IL-8, IL-10, IL-12, TGF-β, TNF-α |
| Treatment-resistant schizophrenia | IL-1β, IL-6, |
| Psychopathology | IL-1β, IL-2, IL-6, IL-17, IL-18, IFN-γ, CRP |
| Cognition | IL-6, CRP |
| Neuroimaging and/or neuroinflammation | IL–6 and hippocampus; TSPO |
a
CRP, C-reactive protein; IFN, interferon; IL, interleukin; R, receptor; RA, receptor antagonist; sIL, soluble interleukin; TGF, transforming growth factor; TNF, tumor necrosis factor; TSPO, translocator protein.
The Immune System and Inflammation
The immune system encompasses two major divisions, the innate and adaptive immune systems. Innate (nonspecific) immunity is the first-line defense against infection and/or cellular injury. Innate immunity provides an immediate, maximal response against a potential pathogen. Whereas it was previously thought that the innate immune system did not elicit immunological memory, increasingly it has been shown that this arm of the immune system may have immunological memory, in that cells may be trained via metabolic and epigenetic modifications, hence referred to as “trained immunity” (23). Important cells of the innate immune system include mononuclear phagocytes (neutrophils, monocytes, and macrophages), which can engulf and destroy pathogens, and natural killer cells, which can destroy virus- or tumor-infected cells. By contrast, the adaptive immune system responds to specific pathogens or antigens (i.e., a foreign particle that triggers antibody production). In response to a foreign antigen, B-lymphocytes are stimulated to produce antibodies to neutralize the antigen, and to create immunologic memory of that antigen, which can produce a rapid response in the case of re-exposure. Antigen presentation also activates T-lymphocytes. CD8 T-lymphocytes can destroy infected or damaged cells. CD4 T-helper (Th) lymphocytes coordinate and maximize adaptive immune responses.
Cytokines are key regulators of inflammation—the complex response of blood vessels to injury, whether caused by infection or cell damage and dysfunction—that coordinate both arms of the immune system. Cytokines are key immune signaling molecules that exert effects by binding specific receptors on a variety of target cells in both the periphery and the brain. Soluble cytokine receptors can either inhibit (e.g., soluble interleukin-2 receptor [sIL-2R]) or enhance (e.g., sIL-6R) the biological activity of cytokines. Endogenous cytokine receptor antagonists (e.g., IL-1 receptor antagonist [IL-1RA]) compete with cytokines for membrane-bound receptors. Cytokines are also key regulators of the acute phase response.
Activated immune cells have characteristic cytokine profiles. In response to an antigen and the local milieu, naïve CD4 Th lymphocytes can differentiate into a variety of effector cells that direct diverse immune functions. Th1 lymphocytes, which secrete interferon (IFN)-γ, IL-2, and IL-12, promote proliferation of CD8 T-lymphocytes and maximize the killing efficacy of macrophages. Th2 lymphocytes secrete IL-4, IL-6, and IL-10, which stimulate proliferation of B-lymphocytes and neutralizing antibody production. Th3 lymphocytes produce transforming growth factor (TGF)-β and IL-10 and are involved in mucosal immunity and protection. Th17 lymphocytes produce IL-17, which mediates inflammation and has been implicated as an effector of autoimmune and other inflammatory disorders. IL-6 induces Th17 cell differentiation and inhibits differentiation of regulatory T cells. Regulatory T cells, which express the transcription factor forkhead box P3, suppress adaptive T-cell responses and prevent autoimmunity. Activated monocytes and/or macrophages enhance the inflammatory response via production of IL-1β, IL-6, IL-8, and tumor necrosis factor (TNF)-α. Concomitant with increased production of pro-inflammatory cytokines, the compensatory anti-inflammatory response syndrome is a counter-regulatory mechanism that inhibits the primary inflammatory response and involves adaptive reprogramming of leukocytes (24). Mediators of the compensatory anti-inflammatory response syndrome include IL-1RA, the soluble TNF receptor (sTNF-R), and TGF-β.
There is significant complexity in regard to how cytokines transmit inflammatory signals from the peripheral immune system to the CNS to drive inflammation in the brain (25). Historically, the brain has largely been considered an immune-privileged organ, although recent evidence has revealed a lymphatic vasculature in the meninges that suggests a link between the peripheral immune system and the CNS, allowing for central immune surveillance (26). Prior to this important finding, the field’s understanding of peripheral-central communication focused on two pathways: humoral and neural. One primary challenge regarding immune signaling to the brain is that inflammatory cytokines are relatively large molecules, which have difficulty crossing the blood-brain barrier (27). The humoral pathway involves cytokines passing through leaky regions of the blood-brain barrier (such as the circumventricular organs) or cytokine binding to transport molecules on the blood-brain barrier. This pathway via the blood-brain barrier may be of specific interest in regard to patients with schizophrenia, who have been shown to have dysfunction of blood-brain barrier integrity and function (28). Alternatively, the neural pathway involves the binding of inflammatory cytokines to peripheral afferent nerve fibers, which, in turn, translates signals into central inflammatory signals. Another more recent finding is that of a third pathway, the cellular pathway, where activated immune cells, primarily monocytes, are trafficked to the brain via chemokines, such as monocyte chemoattractant protein 1, that are produced by microglia, the resident immune cells of the brain (29). Future work to understand relationships between peripheral and central inflammatory markers in schizophrenia will further elucidate the relevance of these mechanisms. Neuroimaging of microglial activation (see below) from patients with schizophrenia, and modeling of microglial signaling from induced pluripotent stem cells from patients with schizophrenia, will further advance our understanding of peripheral-CNS communication.
The brain-gut axis and the microbiome as it relates to the immune system has also been a topic of interest to psychiatry. Microbes that reside in the mucosa of the gastrointestinal tract often have a symbiotic relationship with their host and play a significant role in immune system regulation (30). The microbiome is sensitive to a number of environmental factors and exposures, including diet, infection, stress, and medications. Various genetic factors, including those related to immune genes, place individuals with altered microbiota at risk for immune dysfunction. This altered gut flora, or dysbiosis, can activate the immune system via binding to pattern-recognition receptors associated with microbe-associated molecular patterns, which in turn leads to production of inflammatory cytokines via activation of the nuclear factor kappa-B pathways and the inflammasome (31, 32). Gut microbiota may also communicate with the brain via interactions with the endocrine system (e.g., hypothalamic-pituitary-adrenal axis), the autonomic nervous system, the enteric nervous system, as well as signaling via the vagus nerve (33). Studies of the microbiome in patients with schizophrenia have demonstrated a lack of diversity in gut microbiota, with some evidence for specific bacteria having a relationship with symptom severity (34–36), and there is increasing evidence that altered gut microbiota in patients may be related to immune dysfunction (37, 38). Future directions in this area may involve clinical trials targeting the microbiome to improve dysbiosis and immune alterations in the gastrointestinal system.
Genetic, Prenatal, and Premorbid Inflammation as Risk Factors for Schizophrenia
Genetic Risk Factors
Hundreds of genes have been associated with schizophrenia risk, with most studies having had small effect sizes. Several large genome-wide associated studies have implicated immune genes in the etiopathophysiology of schizophrenia. Polymorphisms in the major histocompatibility complex locus on chromosome 6, which plays a key role in immune system development, are associated with increased schizophrenia risk (3), and the strongest signal within the complex is the complement protein C4 locus (2).
There is also meta-analytic evidence for associations between cytokine gene polymorphisms, including IL1B, IL6, sIL6R, and IL-10, and schizophrenia risk (39–41). These polymorphisms may also have an impact on blood cytokine levels. By contrast, two large Mendelian randomization analyses (42, 43) found that genotypes for the acute phase reactant C-reactive protein (CRP) were associated with higher blood concentrations of CRP, but a significantly decreased risk of schizophrenia. The implications of this discordant finding are unclear, because blood CRP and proinflammatory cytokine levels are positively correlated with each other. Several studies have also investigated blood cytokine levels of first-degree relatives of patients with schizophrenia. There has been replicated evidence for higher blood levels of sIL-2R (44–47) and IL-6 (45, 47) among first-degree relatives, also supporting the possibility of a genetic contribution to inflammation.
Prenatal Inflammation—the Role of Infectious Agents
Prenatal maternal infection with a variety of viruses and bacteria (4), and the parasite Toxoplasma gondii (T. gondii) (48), have been replicated as risk factors for schizophrenia. Given the myriad of infectious agents associated with schizophrenia, the inflammatory response to infection has been posited as a potential common mediator of these associations. Animal models of maternal immune activation with adjuvants in the absence of a specific pathogen—including synthetic double-stranded ribonucleic acid, poly(I:C), which induces an antiviral inflammatory response, or lipopolysaccharide, which mimics bacterial infection, as well as direct activation with cytokines—have been investigated (49). In these animal models, changes in behavior, cognition, brain morphology, cytokine levels, and neurotransmitter function with homology to schizophrenia have been observed among adult offspring.
There is parallel evidence in humans that maternal inflammation during pregnancy is associated with schizophrenia risk. A meta-analysis found that increased maternal blood inflammatory markers, including CRP, IL-8, and IL-10, during pregnancy have been associated with schizophrenia risk (50) for the offspring. A recent study by the National Collaborative Perinatal Project found higher maternal blood TNF-α, IL-1β, and IL-6 levels in offspring who later developed psychosis compared with control subjects, with differences for the first half of pregnancy (51). Importantly, most of these studies measured inflammatory markers at a single, nonstandardized time point during gestation. Prenatal exposure to inflammation is also associated with other abnormalities in immune function, neurocognition, brain morphology, and gene expression in adults with schizophrenia (52). These findings support the hypothesis that prenatal inflammation may alter fetal brain development, thereby increasing vulnerability to schizophrenia.
Premorbid Inflammation
The association between inflammation and schizophrenia risk also appears to extend beyond the prenatal period, to include the neonatal period, childhood, and adolescence. A study that used newborn dried blood spot testing found increased complement C4 levels in newborns who later developed schizophrenia (53). Paradoxically, another study found that decreased levels of neonatal acute phase proteins on dried blood spot tests were associated with significantly increased risk of psychosis (54), as well as a potential interaction between maternal infection with T. gondii or cytomegalovirus and low acute phase protein levels on schizophrenia risk (55). Hospitalization for autoimmune disorders and/or infections during childhood or adolescence, both of which are associated with increased inflammation, is associated with schizophrenia (6), and schizophrenia is also a risk factor for having autoimmune disorders and infections (5). Furthermore, the presence of an autoimmune disorder and hospitalization for infections may act synergistically on subsequent schizophrenia risk. A cohort study found that blood IL-6 levels at age 9 were associated with risk of psychotic experiences and fulminant psychotic disorder at age 18 (56). Higher IL-6 levels during childhood were associated with risk of psychotic experiences in a dose-dependent fashion. In this cohort, a genetic variant of sIL6R (which decreases inflammation) was associated with decreased risk of psychosis, increased blood IL-6 levels, and decreased blood CRP levels (57).
Inflammation Among Patients With Clinical High Risk (CHR) for Psychosis
Investigation of inflammation in individuals at CHR for psychosis is important because it represents a potential diagnostic, prognostic, and theranostic marker. The population of individuals at CHR is heterogeneous, and current estimations suggest that approximately 20% will experience a first-episode of psychosis (FEP) within 2 years (58). A recent meta-analysis of seven studies found that blood IL-6 levels were significantly increased and IL-1β levels were significantly decreased among individuals with CHR compared with healthy control subjects (59). In this meta-analysis (59), there was a trend for increased blood IL-12 levels among subjects who developed (versus those who did not develop) FEP. One study found that blood IL-6 levels were positively associated with both positive and negative symptoms, as measured by the Positive and Negative Syndrome Scale (PANSS), among young individuals with at-risk mental states (60). Another study (61) found a positive association between higher TNF-α and worsening negative symptom trajectories, as well as an association between lower IL-6 and worsening negative symptom trajectories, among individuals from the North American Prodromal Longitudinal Study Cohort. Despite a limited number of studies, there is evidence for alterations in inflammatory markers of some individuals with CHR, which may be relevant to development of psychosis and may be related to some domains of psychopathology. It seems plausible that those individuals at CHR who have altered inflammatory markers in the illness prodrome may be those who subsequently develop inflammation in FEP and chronic schizophrenia.
Autoimmunity in Schizophrenia
There is increased prevalence of several autoimmune diseases—which are associated with inflammation—among patients with schizophrenia (62, 63) and other nonaffective psychoses (64), as well as among their first-degree relatives (63, 64), compared with control subjects. These autoimmune disorders include acquired hemolytic anemia, celiac disease, interstitial cystitis, Sjogren’s syndrome, and thyrotoxicosis (63, 64). The association between schizophrenia and autoimmune disorders appears to be bidirectional. Both a personal (63) as well as a family history (64) of any autoimmunity is associated with a significantly increased risk of schizophrenia. Conversely, patients with schizophrenia have an increased risk of incident autoimmune disease, and a history of hospitalization for infection acts synergistically on this risk (6).
A meta-analysis of 81 studies (19) found significantly increased prevalence of positive titers for 20 different autoantibodies among patients with schizophrenia compared with control subjects. Absolute titers of anticardiolipin immunoglobulin G (IgG) and M (IgM), and nerve growth factor antibodies were significantly increased among the patients with schizophrenia. Lachance and McKenzie (65) performed a meta-analysis of 12 studies of biomarkers of gluten sensitivity among patients with schizophrenia, finding a significant increase in antigliadin IgG, anti-tissue transglutaminase 2 IgA, and anti-wheat antibodies, suggesting an immune response that differs from patients with celiac disease. A recent study of patients with FEP from China (66) found higher levels of anticytokine antibodies, including anti-IL-1β, anti-IL-6, and anti-IL-8 IgG. Other meta-analyses (67) have found an increase in anti-NMDA receptor antibodies among patients with schizophrenia compared with control subjects that is based on high- but not low-specificity seropositivity thresholds. Indeed, these cases of autoimmune encephalitis caused by anti-NMDA receptor antibodies may masquerade as new-onset psychosis. These findings lead to the question of what is the pathophysiologic significance of such autoantibodies among patients with schizophrenia, particularly in the absence of other signs and symptoms of autoimmune disease, and whether these autoantibodies might be targets for novel immunotherapy approaches (e.g., corticosteroids or plasmapheresis).
Inflammation in First-Episode Psychosis
In a meta-analysis of patients with FEP and chronic psychosis (68), many inflammatory markers were elevated, including IFN-γ, IL-1β, IL-1RA, IL-6, IL-8, IL-10, IL-12, sIL-2R, TGF-β, and TNF-α. There was similar directionality between patients with FEP and chronic schizophrenia for IL-1β, IL-6, sIL-2R, and TNF-α. In another meta-analysis of studies of antipsychotic-naïve patients with FEP (69), IL-1β, sIL-2R, IL-6, and TNF-α were elevated compared with healthy control subjects, suggesting associations with inflammation that were unrelated to antipsychotic medications. Ultimately more longitudinal data will be necessary to appreciate changes in inflammatory markers over the course of illness in psychosis.
Inflammation in Treatment-Resistant Psychosis
Compared with other patient populations, there have been substantially fewer studies of inflammation of patients with treatment-resistant psychosis. There has been evidence for higher IL-1β and IL-6 levels among clozapine-treated patients versus control subjects in at least two studies, although there have been failures to replicate the results in some studies (70–74). There have been even fewer longitudinal studies of inflammatory markers among patients before and after initiation of clozapine. Several small studies (75–78) have reported changes in sIL-2R and IL-6 during the initial weeks of clozapine treatment, but the findings have been heterogeneous, with the absence of longer-term data. One study (79) found that baseline and changes in IL-6 levels were significantly positively correlated with psychopathology scores following clozapine treatment. The effects of clozapine on other inflammatory markers, however, have been inconsistent (75, 78, 80–83). Given that there are no established blood-based markers that predict response to clozapine, this finding represents an important potential area for future investigations.
Inflammation, Psychopathology, and Infections in Schizophrenia
Inflammation and Psychopathology in Schizophrenia
There is evidence for a dose-dependent effect of inflammation on psychopathology in patients with schizophrenia. In a meta-analysis of 73 studies (84), encompassing 6,112 patients, CRP was positively correlated with total psychopathology and negative symptoms, and IL-18 was positively correlated with total and general psychopathology. Furthermore, among inpatients with schizophrenia, IL-6, CRP, IL-17, IL-18, and IFN-γ were all correlated with multiple domains of psychopathology. In patients with FEP, IL-1β and IL-2 were correlated with total psychopathology. Following antipsychotic treatment, changes in IL-6 levels were also correlated with changes in total psychopathology.
Findings from studies of the general population have shown that increased inflammation is associated with alterations in cognition, including memory and executive function. There has been modest evidence for associations between higher blood IL-6 and CRP levels and greater cognitive impairment among patients with FEP and chronic schizophrenia, including executive function, verbal memory and learning, and psychomotor speed, although there have been failures to replicate these findings (85–87). Another recent study (88) found that among patients with psychosis, higher IL-6 levels were associated with larger brain choroid plexus volume, which in turn, were associated with worse cognition. A recent clinical trial of adjunctive treatment with the tetracycline antibiotic minocycline, which exerts anti-inflammatory effects via inhibition of brain microglia, found that improvements in cognition among patients with schizophrenia were correlated with reductions in blood IL-6 levels (89). Taken together, these findings suggest that there may be a biological gradient to the association between inflammation and psychopathology in schizophrenia.
Infections and Acute Psychosis
Schizophrenia is also associated with comorbid infections. There is increased prevalence of urinary tract infections (UTI) among patients with schizophrenia, particularly during psychotic illness exacerbations (90), which may be a recurrent phenomenon (91). A meta-analysis (92) also found a 1.7-fold increase in positive T. gondii IgM antibodies—a marker of acute and/or recent exposure or reinfection—among patients with acute psychosis, compared with control subjects. Several other studies have found increased prevalence of active viral (93, 94) and chlamydial (95) infections among hospitalized patients with acute psychosis. An important limitation of these studies is that they do not permit inferences regarding temporal or causal aspects of the association with psychosis. However, a recent population-based sample from Denmark (96) found an association between genetic susceptibility to infection and schizophrenia.
Imaging of Neuroinflammation in Schizophrenia
There is evidence of associations between peripheral blood inflammation, namely IL-6, and smaller hippocampal volumes among patients with chronic schizophrenia (97, 98) and FEP (99, 100). A recent study (101) found that among outpatients with schizophrenia, increased blood levels of IL-6, IL-8, and IL-10 were significantly associated with cortical thickness measurements in the orbital frontal cortex, frontotemporal gyrus, and cingulate cortex. In diffusion tensor imaging, higher blood IL-6 levels have also been negatively correlated with fractional anisotropy and positively correlated with radial diffusion in patients with schizophrenia (102). In a study of patients with FEP (103), the percentage of whole-brain gray matter was inversely correlated with IFN-γ and IL-12 levels. Another small study of outpatients with schizophrenia and elevated cytokine mRNA levels (104) showed a significant reduction in the left pars opercularis (Broca’s area), as well as a negative relationship between IL-1β mRNA levels and left pars opercularis volume. In the North American Prodrome Longitudinal Study (105), the rate of prefrontal cortical thinning was significantly associated with higher baseline levels of proinflammatory cytokines, and this inverse correlation was significantly greater among subjects who developed a psychotic disorder than among those who did not and among control subjects. Two studies (106, 107) have also found significantly increased IL-6 mRNA expression in the prefrontal cortex of patients with schizophrenia. A third postmortem transcriptional study (108) found upregulated gene expression of the IL-6 signaling pathway in the hippocampus, prefrontal cortex, and striatum among patients with schizophrenia compared with control subjects.
Whereas the investigation of inflammation in schizophrenia has largely focused on measurement of peripheral blood markers, there is also significant evidence for increased density and activation of microglia in the brain, reflective of neuroinflammation, that has been demonstrated in both neuropathological and neuroimaging studies (109–111). Imaging of microglial activation using positron emission tomography (PET) is a burgeoning field with an increasing number of studies. As the resident immune cells of the brain, microglia release cytokines and other molecular mediators to promote an inflammatory response (M1 phenotype) while simultaneously downgrading the anti-inflammatory response (M2 phenotype) (112). When activated, microglia express translocator protein (TSPO), and TSPO radioligands have been developed for PET imaging (113). The findings from these TSPO studies have been mixed. In an individual participant meta-analysis of five studies of second-generation TSPO radioligands, Plavén-Sigray and colleagues (114) showed that individuals with FEP and schizophrenia had lower levels of TSPO (as measured by total distribution volume [VT]), compared with healthy control subjects, in the frontal cortex, temporal cortex, and hippocampus. A second, recent meta-analysis of 12 TSPO PET studies showed that binding potential was elevated among patients with schizophrenia compared with control subjects, despite no difference in VT (115). The discrepancies between these meta-analyses highlight the distinct challenges in interpreting these studies of neuroinflammation.
Inflammation and Treatment in Schizophrenia
Inflammatory Biomarkers of Treatment Response in Schizophrenia
Given the evidence for cytokine alterations in patients with schizophrenia, these markers may represent a potential predictor of treatment response. Indeed, one study (116) found that patients with FEP who did not respond to treatment had higher baseline IL-6 and IFN-γ levels compared with patients who responded after 12 weeks of antipsychotic treatment, and concentrations of both cytokines remained elevated among the nonresponders. More recent work from the same researchers (117) showed that the combination of the CRP and metabolic markers—body mass index and triglycerides—predicted worse treatment response after 1 year. Moreover, this combined measure of metabolic-inflammatory status predicted worse negative symptoms at baseline, and worse severity of total, positive, and general psychopathology at the 1-year follow-up. This group also found that high baseline complement C4 predicted worse clinical outcome (treatment nonresponse) at 1-year follow-up in patients with FEP (118). By contrast, a recent study (119) of acutely ill patients with FEP or chronic schizophrenia found that higher baseline blood IL-6 predicted a decrease in PANSS negative symptoms at 6 months. Thus, future studies are needed regarding the potential of inflammatory markers as predictors of treatment response among patients with schizophrenia.
Inflammation as a Therapeutic Target in Schizophrenia
Two meta-analyses have explored the efficacy of adjunctive anti-inflammatory agents in schizophrenia. Nitta et al. (12) performed a meta-analysis of eight studies of adjunctive NSAIDs (six trials of celecoxib and two trials of aspirin), finding that NSAIDs were associated with a small, trend-level reduction in PANSS total score. Associations for improvements with adjunctive NSAIDs were stronger among inpatients with schizophrenia and those with FEP compared to other patient groups. These findings are broadly consistent with meta-analytic findings of associations between abnormal inflammatory markers and clinical status (68, 120). In another meta-analysis of 26 double-blind trials of various anti-inflammatory agents, Sommer et al. (13) found significant effects for aspirin, estrogens, and N-acetylcysteine. These heterogeneous findings are consistent with the hypothesis that inflammation may be present in only a subset of patients with schizophrenia, which may influence the likelihood of response. Accordingly, two trials (14, 15) found that baseline blood cytokine levels predicted response to adjunctive NSAIDs among patients with schizophrenia.
Important limitations of trials of adjunctive NSAIDs are that these agents have relevant off-target (i.e., nonimmune) effects and have less potent anti-inflammatory properties than other immunomodulators, such as monoclonal antibodies. Many humanized monoclonal antibodies have been approved for the treatment of autoimmune disorders and cancers. These agents act by directly neutralizing cytokines or cytokine receptors, and therefore offer an unparalleled opportunity to directly test for a pathophysiological role for inflammation in schizophrenia. To date, there are four published studies of cytokine-based immunotherapy for patients with schizophrenia, although several larger trials are ongoing. Grüber et al. (121) presented a case series of two patients with treatment-resistant schizophrenia who had significant improvement in total psychopathology with adjunctive recombinant human IFN-γ-1b. Another study (122) reported on an 8-week open-label trial of adjunctive tocilizumab (an anti-interleukin-6 receptor monoclonal antibody) in six patients with schizophrenia, finding significant improvements in verbal fluency, digit symbol coding, and global cognition. However, a recent randomized-controlled trial (123) of adjunctive tocilizumab did not improve psychopathology or cognition in patients with chronic schizophrenia. An 8-week trial (124) of the IL-1β antagonist canakinumab found a significant reduction in positive symptoms at study endpoint, with a trend-level correlation between reduction in CRP and decreased positive symptoms. Findings from ongoing trials of adjunctive monoclonal antibody immunotherapy in schizophrenia—many of which have required elevated peripheral blood inflammatory markers for inclusion—regardless of outcome, will provide valuable insights into the role of inflammation in schizophrenia.
Underlying Mechanisms
Figure 1 presents a theoretical framework that integrates findings regarding potential mechanistic associations between inflammation and schizophrenia. Briefly, abnormal allostasis interacts with genetic and/or epigenetic factors, which results in peripheral inflammation, characterized by cellular activation, cytokine production, and the acute phase response. In the setting of increased blood-brain barrier permeability, there is also central inflammation, including cytokine production, microglial activation, and activation of the tryptophan catabolic pathway. As a result, inflammation can directly or indirectly affect a variety of processes, including brain connectivity, neurodevelopment, neurogenesis, neurotransmitter function, and white matter function, thereby contributing to schizophrenia psychopathology. For example, increased blood IL-6 in rodents directly modulates dopamine turnover and sensitization to amphetamine-induced locomotion (125). By contrast, the activity of indoleamine 2,3-dioxygenase (IDO), the rate-limiting enzyme in tryptophan catabolism that is expressed in microglia, can be modulated by cytokines. IDO induction results in increased production of kynurenine, which is converted into the NMDA receptor antagonist kynurenic acid, and this pathway has been implicated in schizophrenia (126). Inflammation may also play a role in specific symptom domains, such as negative symptoms and cognitive impairment.
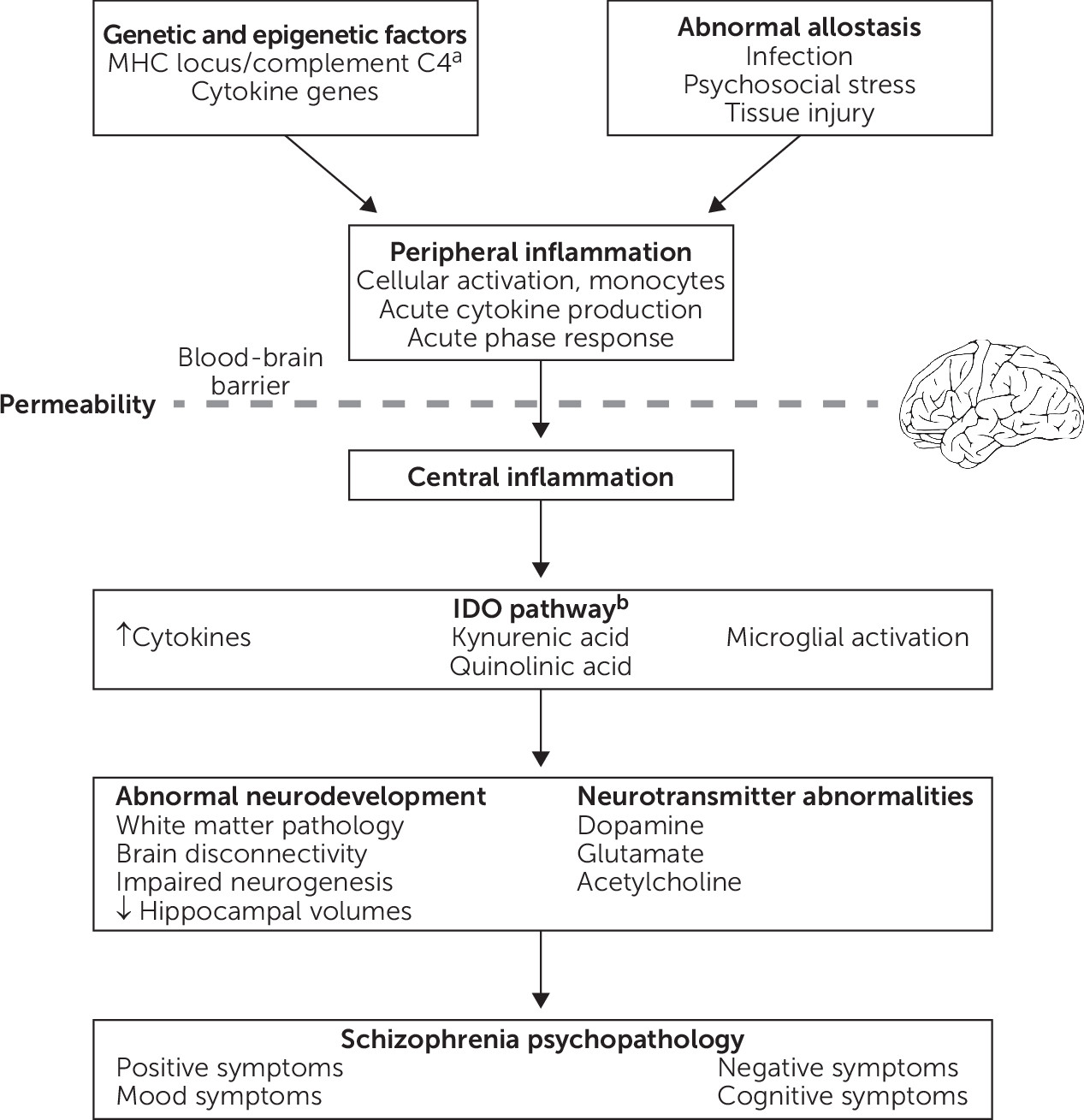
FIGURE 1. Potential mechanisms underlying associations between inflammation and schizophreniaa
aMHC, major histocompatibility complex. bIDO, indoleamine 2,3-dioxygenase.
Conclusions and Future Research
The decades-long literature on inflammation in schizophrenia is fraught with significant heterogeneity, including negative studies. Many associations between inflammation and schizophrenia have had small-to-moderate effect sizes. Possible explanations for heterogeneous findings include minor inflammation-associated changes that may not be clinically significant, study-related factors (e.g., small sample size and inadequate consideration of potential confounding and/or moderating factors), and/or inflammation that occurs among only a subset of patients with schizophrenia. Regarding the latter, a recent meta-analysis of the variability and distribution of cytokines in FEP did not find evidence for an immune subgroup (127). In contrast, associations between baseline blood cytokine levels and response to adjunctive NSAID treatment among patients with schizophrenia (14, 15) raise the possibility of a subgroup of patients for whom inflammation may be a marker of treatment response. Each of these hypotheses warrants further investigation.
Despite substantial heterogeneity, there is replicated evidence for inflammation in schizophrenia in multiple domains, including risk factors, clinical course of illness, and symptomatology (Table 1). Thus far, inflammatory markers with replicated positive findings across multiple domains include IL-1β, IL-6, and CRP. Although there is evidence for changes in levels of inflammatory markers with antipsychotic treatment (68, 120), there has been less investigation of inflammation as a marker of response to treatment in schizophrenia. Studies of specific anti-inflammatory strategies, such as monoclonal antibody immunotherapy, are also in their relative infancy. Furthermore, a small number of studies have afforded evidence for a potential causal association between inflammation and schizophrenia. For example, the study by Sekar et al. (2) has provided compelling evidence that the C4 gene may increase schizophrenia risk by influencing synaptic pruning during critical periods of brain development. In mice, a single maternal injection of IL-6 during pregnancy caused prepulse and latent inhibition deficits in the adult offspring, suggesting the immune response may mediate robust epidemiologic evidence for the association between prenatal maternal infections and schizophrenia risk in the offspring (128).
In conclusion, inflammation likely plays an important role in the pathology of schizophrenia for at least some patients, although there is not clear evidence to support the assertion that schizophrenia is truly an inflammatory disorder. This area of study shows promise toward a greater understanding of the etiopathophysiology of this heterogeneous syndrome and toward identification of potential illness subtypes, diagnostics, and therapeutics. These efforts aim to reduce illness risk, alleviate symptoms, advance relapse prevention efforts, inform treatment effectiveness, and affect disease progression, ultimately toward more personalized medicine approaches and improved quality of life for at-risk and patients with established schizophrenia.
References
1.
Purcell SM, Wray NR, Stone JL, et al: Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460:748–752
2.
Sekar A, Bialas AR, de Rivera H, et al: Schizophrenia risk from complex variation of complement component 4. Nature 2016; 530:177–183
3.
Shi J, Levinson DF, Duan J, et al: Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009; 460:753–757
4.
Brown AS, Derkits EJ: Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry 2010; 167:261–280
5.
Benros ME, Pedersen MG, Rasmussen H, et al: A nationwide study on the risk of autoimmune diseases in individuals with a personal or a family history of schizophrenia and related psychosis. Am J Psychiatry 2014; 171:218–226
6.
Nielsen PR, Benros ME, Mortensen PB: Hospital contacts with infection and risk of schizophrenia: a population-based cohort study with linkage of Danish national registers. Schizophr Bull 2014; 40:1526–1532
7.
Kirkpatrick B, Miller BJ: Inflammation and schizophrenia. Schizophr Bull 2013; 39:1174–1179
8.
Mazza MG, Lucchi S, Rossetti A, et al: Neutrophil-lymphocyte ratio, monocyte-lymphocyte ratio and platelet-lymphocyte ratio in non-affective psychosis: a meta-analysis and systematic review. World J Biol Psychiatry 2020; 21:326–338
9.
Miller BJ, Culpepper N, Rapaport MH: C-reactive protein levels in schizophrenia: a review and meta-analysis. Clin Schizophr Relat Psychoses 2014; 7:223–230
10.
Wang AK, Miller BJ: Meta-analysis of cerebrospinal fluid cytokine and tryptophan catabolite alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder, and depression. Schizophr Bull 2018; 44:75–83
11.
Chae J, Miller B: Beyond urinary tract infections (UTIs) and delirium: a systematic review of UTIs and neuropsychiatric disorders. J Psychiatr Pract 2015; 21:402–411
12.
Nitta M, Kishimoto T, Müller N, et al: Adjunctive use of nonsteroidal anti-inflammatory drugs for schizophrenia: a meta-analytic investigation of randomized controlled trials. Schizophr Bull 2013; 39:1230–1241
13.
Sommer IE, van Westrhenen R, Begemann MJ, et al: Efficacy of anti-inflammatory agents to improve symptoms in patients with schizophrenia: an update. Schizophr Bull 2014; 40:181–191
14.
Laan W, Grobbee DE, Selten JP, et al: Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2010; 71:520–527
15.
Müller N, Ulmschneider M, Scheppach C, et al: COX-2 inhibition as a treatment approach in schizophrenia: immunological considerations and clinical effects of celecoxib add-on therapy. Eur Arch Psychiatry Clin Neurosci 2004; 254:14–22
16.
Sommer IE, van Bekkum DW, Klein H, et al: Severe chronic psychosis after allogeneic SCT from a schizophrenic sibling. Bone Marrow Transplant 2015; 50:153–154
17.
Miyaoka T, Wake R, Hashioka S, et al: Remission of psychosis in treatment-resistant schizophrenia following bone marrow transplantation: a case report. Front Psychiatry 2017; 8:174
18.
Dickerson F, Severance E, Yolken R: The microbiome, immunity, and schizophrenia and bipolar disorder. Brain Behav Immun 2017; 62:46–52
19.
Ezeoke A, Mellor A, Buckley P, et al: A systematic, quantitative review of blood autoantibodies in schizophrenia. Schizophr Res 2013; 150:245–251
20.
Flatow J, Buckley P, Miller BJ: Meta-analysis of oxidative stress in schizophrenia. Biol Psychiatry 2013; 74:400–409
21.
Miller BJ, Goldsmith DR: Towards an immunophenotype of schizophrenia: progress, potential mechanisms, and future directions. Neuropsychopharmacology 2017; 42:299–317
22.
Woo JJ, Pouget JG, Zai CC, et al: The complement system in schizophrenia: where are we now and what’s next? Mol Psychiatry 2020; 25:114–130
23.
Netea MG, Domínguez-Andrés J, Barreiro LB, et al: Defining trained immunity and its role in health and disease. Nat Rev Immunol 2020; 20:375–388
24.
Adib-Conquy M, Cavaillon JM: Compensatory anti-inflammatory response syndrome. Thromb Haemost 2009; 101:36–47
25.
Miller AH, Raison CL: The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 2016; 16:22–34
26.
Louveau A, Herz J, Alme MN, et al: CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci 2018; 21:1380–1391
27.
Quan N, Banks WA: Brain-immune communication pathways. Brain Behav Immun 2007; 21:727–735
28.
Pollak TA, Drndarski S, Stone JM, et al: The blood-brain barrier in psychosis. Lancet Psychiatry 2018; 5:79–92
29.
D’Mello C, Le T, Swain MG: Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci 2009; 29:2089–2102
30.
Golofast B, Vales K: The connection between microbiome and schizophrenia. Neurosci Biobehav Rev 2020; 108:712–731
31.
Takeuchi O, Akira S: Pattern recognition receptors and inflammation. Cell 2010; 140:805–820
32.
Fleshner M: Stress-evoked sterile inflammation, danger associated molecular patterns (DAMPs), microbial associated molecular patterns (MAMPs) and the inflammasome. Brain Behav Immun 2013; 27:1–7
33.
Kim YK, Shin C: The microbiota-gut-brain axis in neuropsychiatric disorders: pathophysiological mechanisms and novel treatments. Curr Neuropharmacol 2018; 16:559–573
34.
Schwarz E, Maukonen J, Hyytiäinen T, et al: Analysis of microbiota in first episode psychosis identifies preliminary associations with symptom severity and treatment response. Schizophr Res 2018; 192:398–403
35.
Shen Y, Xu J, Li Z, et al: Analysis of gut microbiota diversity and auxiliary diagnosis as a biomarker in patients with schizophrenia: a cross-sectional study. Schizophr Res 2018; 197:470–477
36.
Yuan X, Zhang P, Wang Y, et al: Changes in metabolism and microbiota after 24-week risperidone treatment in drug naïve, normal weight patients with first episode schizophrenia. Schizophr Res 2018; 201:299–306
37.
Severance EG, Gressitt KL, Stallings CR, et al: Discordant patterns of bacterial translocation markers and implications for innate immune imbalances in schizophrenia. Schizophr Res 2013; 148:130–137
38.
Yolken RH, Severance EG, Sabunciyan S, et al: Metagenomic sequencing indicates that the oropharyngeal phageome of individuals with schizophrenia differs from that of controls. Schizophr Bull 2015; 41:1153–1161
39.
Gao L, Li Z, Chang S, et al: Association of interleukin-10 polymorphisms with schizophrenia: a meta-analysis. PLoS One 2014; 9:e90407
40.
Hudson ZD, Miller BJ: Meta-analysis of cytokine and chemokine genes in schizophrenia. Clin Schizophr Relat Psychoses 2018; 12:121–129B
41.
Shibuya M, Watanabe Y, Nunokawa A, et al: Interleukin 1 beta gene and risk of schizophrenia: detailed case-control and family-based studies and an updated meta-analysis. Hum Psychopharmacol 2014; 29:31–37
42.
Ligthart S, Vaez A, Võsa U, et al: Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. Am J Hum Genet 2018; 103:691–706
43.
Prins BP, Abbasi A, Wong A, et al: Investigating the causal relationship of c-reactive protein with 32 complex somatic and psychiatric outcomes: a large-scale cross-consortium Mendelian randomization study. PLoS Med 2016; 13:e1001976
44.
Gaughran F, O’Neill E, Sham P, et al: Soluble interleukin 2 receptor levels in families of people with schizophrenia. Schizophr Res 2002; 56:235–239
45.
Martínez-Gras I, García-Sánchez F, Guaza C, et al: Altered immune function in unaffected first-degree biological relatives of schizophrenia patients. Psychiatry Res 2012; 200:1022–1025
46.
Rapaport MH, Torrey EF, McAllister CG, et al: Increased serum soluble interleukin-2 receptors in schizophrenic monozygotic twins. Eur Arch Psychiatry Clin Neurosci 1993; 243:7–10
47.
Nunes SO, Matsuo T, Kaminami MS, et al: An autoimmune or an inflammatory process in patients with schizophrenia, schizoaffective disorder, and in their biological relatives. Schizophr Res 2006; 84:180–182
48.
Torrey EF, Bartko JJ, Lun ZR, et al: Antibodies to Toxoplasma gondii in patients with schizophrenia: a meta-analysis. Schizophr Bull 2007; 33:729–736
49.
Meyer U: Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry 2014; 75:307–315
50.
Zhang J, Luo W, Huang P, et al: Maternal C-reactive protein and cytokine levels during pregnancy and the risk of selected neuropsychiatric disorders in offspring: a systematic review and meta-analysis. J Psychiatr Res 2018; 105:86–94
51.
Allswede DM, Yolken RH, Buka SL, et al: Cytokine concentrations throughout pregnancy and risk for psychosis in adult offspring: a longitudinal case-control study. Lancet Psychiatry 2020; 7:254–261.
52.
Miller BJ, Culpepper N, Rapaport MH, et al: Prenatal inflammation and neurodevelopment in schizophrenia: a review of human studies. Prog Neuropsychopharmacol Biol Psychiatry 2013a; 42:92–100
53.
Cooper JD, Ozcan S, Gardner RM, et al: Schizophrenia-risk and urban birth are associated with proteomic changes in neonatal dried blood spots. Transl Psychiatry 2017; 7:1290
54.
Gardner RM, Dalman C, Wicks S, et al: Neonatal levels of acute phase proteins and later risk of non-affective psychosis. Transl Psychiatry 2013; 3:e228
55.
Blomström Å, Gardner RM, Dalman C, et al: Influence of maternal infections on neonatal acute phase proteins and their interaction in the development of non-affective psychosis. Transl Psychiatry 2015; 5:e502
56.
Khandaker GM, Pearson RM, Zammit S, et al: Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study. JAMA Psychiatry 2014; 71:1121–1128
57.
Khandaker GM, Zammit S, Burgess S, et al: Association between a functional interleukin 6 receptor genetic variant and risk of depression and psychosis in a population-based birth cohort. Brain Behav Immun 2018; 69:264–272
58.
Fusar-Poli P, Cappucciati M, Borgwardt S, et al: Heterogeneity of psychosis risk within individuals at clinical high risk: a meta-analytical stratification. JAMA Psychiatry 2016; 73:113–120
59.
Park S, Miller BJ: Meta-analysis of cytokine and C-reactive protein levels in high-risk psychosis. Schizophr Res 2019; doi:
60.
Stojanovic A, Martorell L, Montalvo I, et al: Increased serum interleukin-6 levels in early stages of psychosis: associations with at-risk mental states and the severity of psychotic symptoms. Psychoneuroendocrinology 2014; 41:23–32
61.
Goldsmith DR, Haroon E, Miller AH, et al: Association of baseline inflammatory markers and the development of negative symptoms in individuals at clinical high risk for psychosis. Brain Behav Immun 2019; 76:268–274
62.
Benros ME, Nielsen PR, Nordentoft M, et al: Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am J Psychiatry 2011; 168:1303–1310
63.
Eaton WW, Byrne M, Ewald H, et al: Association of schizophrenia and autoimmune diseases: linkage of Danish national registers. Am J Psychiatry 2006; 163:521–528
64.
Eaton WW, Pedersen MG, Nielsen PR, et al: Autoimmune diseases, bipolar disorder, and non-affective psychosis. Bipolar Disord 2010; 12:638–646
65.
Lachance LR, McKenzie K: Biomarkers of gluten sensitivity in patients with non-affective psychosis: a meta-analysis. Schizophr Res 2014; 152:521–527
66.
Liu S, Zhang X, Wang J, et al: Analysis of plasma autoantibodies for inflammatory cytokines in patients with first-episode schizophrenia among a Chinese population. J Neuroimmunol 2020; 341:577165
67.
Pollak TA, McCormack R, Peakman M, et al: Prevalence of anti-N-methyl-D-aspartate (NMDA) receptor [corrected] antibodies in patients with schizophrenia and related psychoses: a systematic review and meta-analysis. Psychol Med 2014; 44:2475–2487
68.
Goldsmith DR, Rapaport MH, Miller BJ: A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry 2016; 21:1696–1709
69.
Upthegrove R, Manzanares-Teson N, Barnes NM: Cytokine function in medication-naive first episode psychosis: a systematic review and meta-analysis. Schizophr Res 2014; 155:101–108
70.
Chen DC, Qi LY, Xiu MH, et al: Elevated serum levels of tumor necrosis factor-alpha in clozapine-associated obesity in chronic schizophrenia. Schizophr Res 2008; 106:367–368
71.
Haack M, Hinze-Selch D, Fenzel T, et al: Plasma levels of cytokines and soluble cytokine receptors in psychiatric patients upon hospital admission: effects of confounding factors and diagnosis. J Psychiatr Res 1999; 33:407–418
72.
Klemettilä JP, Kampman O, Seppälä N, et al: Cytokine and adipokine alterations in patients with schizophrenia treated with clozapine. Psychiatry Res 2014; 218:277–283
73.
O’Connell KE, Thakore J, Dev KK: Pro-inflammatory cytokine levels are raised in female schizophrenia patients treated with clozapine. Schizophr Res 2014; 156:1–8
74.
Schmitt A, Bertsch T, Tost H, et al: Increased serum interleukin-1beta and interleukin-6 in elderly, chronic schizophrenic patients on stable antipsychotic medication. Neuropsychiatr Dis Treat 2005; 1:171–177
75.
Kluge M, Schuld A, Schacht A, et al: Effects of clozapine and olanzapine on cytokine systems are closely linked to weight gain and drug-induced fever. Psychoneuroendocrinology 2009; 34:118–128
76.
Löffler S, Klimke A, Kronenwett R, et al: Clozapine mobilizes CD34+ hematopoietic stem and progenitor cells and increases plasma concentration of interleukin 6 in patients with schizophrenia. J Clin Psychopharmacol 2010; 30:591–595
77.
Maes M, Bosmans E, Kenis G, et al: In vivo immunomodulatory effects of clozapine in schizophrenia. Schizophr Res 1997; 26:221–225
78.
Pollmächer T, Hinze-Selch D, Mullington J: Effects of clozapine on plasma cytokine and soluble cytokine receptor levels. J Clin Psychopharmacol 1996; 16:403–409
79.
Lü LX, Guo SQ, Chen W, et al: Effect of clozapine and risperidone on serum cytokine levels in patients with first-episode paranoid schizophrenia. [in Chinese] J First Mil Med Univ 2004; 24:1251–1254
80.
Ajami A, Abedian F, Hamzeh Hosseini S, et al: Serum TNF-α, IL-10 and IL-2 in schizophrenic patients before and after treatment with risperidone and clozapine. Iran J Immunol 2014; 11:200–209
81.
Löffler S, Löffler-Ensgraber M, Fehsel K, et al: Clozapine therapy raises serum concentrations of high sensitive C-reactive protein in schizophrenic patients. Int Clin Psychopharmacol 2010; 25:101–106
82.
Maes M, Meltzer HY, Bosmans E: Immune-inflammatory markers in schizophrenia: comparison to normal controls and effects of clozapine. Acta Psychiatr Scand 1994; 89:346–351
83.
Monteleone P, Fabrazzo M, Tortorella A, et al: Plasma levels of interleukin-6 and tumor necrosis factor alpha in chronic schizophrenia: effects of clozapine treatment. Psychiatry Res 1997; 71:11–17
84.
Miller B, Essali N: Meta-analysis of cytokine levels and psychopathology in schizophrenia. Schizophr Bull 2019; 45:S191–S192
85.
Miller B, Mellor A, Buckley PF: Interleukin-6 and cognition in non-affective psychosis. Schizophr Bull 2013; 39:S242–S243
86.
Misiak B, Stańczykiewicz B, Kotowicz K, et al: Cytokines and C-reactive protein alterations with respect to cognitive impairment in schizophrenia and bipolar disorder: a systematic review. Schizophr Res 2018; 192:16–29
87.
Ribeiro-Santos R, de Campos-Carli SM, Ferretjans R, et al: The association of cognitive performance and IL-6 levels in schizophrenia is influenced by age and antipsychotic treatment. Nord J Psychiatry 2020; 74:187–193
88.
Lizano P, Lutz O, Ling G, et al: Association of choroid plexus enlargement with cognitive, inflammatory, and structural phenotypes across the psychosis spectrum. Am J Psychiatry 2019; 176:564–572
89.
Zhang L, Zheng H, Wu R, et al: The effect of minocycline on amelioration of cognitive deficits and pro-inflammatory cytokines levels in patients with schizophrenia. Schizophr Res 2019; 212:92–98
90.
Miller BJ, Graham KL, Bodenheimer CM, et al: A prevalence study of urinary tract infections in acute relapse of schizophrenia. J Clin Psychiatry 2013c; 74:271–277
91.
Laney D, Philip N, Miller BJ: Recurrent urinary tract infections in acute psychosis. Schizophr Res 2015; 164:275–276
92.
Monroe JM, Buckley PF, Miller BJ: Meta-analysis of anti-Toxoplasma gondii IgM antibodies in acute psychosis. Schizophr Bull 2015; 41:989–998
93.
Ahokas A, Rimón R, Koskiniemi M, et al: Viral antibodies and interferon in acute psychiatric disorders. J Clin Psychiatry 1987; 48:194–196
94.
Srikanth S, Ravi V, Poornima KS, et al: Viral antibodies in recent onset, nonorganic psychoses: correspondence with symptomatic severity. Biol Psychiatry 1994; 36:517–521
95.
Fellerhoff B, Laumbacher B, Mueller N, et al: Associations between Chlamydophila infections, schizophrenia and risk of HLA-A10. Mol Psychiatry 2007; 12:264–272
96.
Nudel R, Wang Y, Appadurai V, et al: A large-scale genomic investigation of susceptibility to infection and its association with mental disorders in the Danish population. Transl Psychiatry 2019; 9:283
97.
Miller BJ, Herzig KH, Jokelainen J, et al: Inflammation, hippocampal volume, and cognition in schizophrenia: results from the Northern Finland 1966 Birth Cohort. Eur Arch Psychiatry Clin Neuroscience 2020; doi:
98.
Kalmady SV, Venkatasubramanian G, Shivakumar V, et al: Relationship between interleukin-6 gene polymorphism and hippocampal volume in antipsychotic-naïve schizophrenia: evidence for differential susceptibility? PLoS One 2014; 9:e96021
99.
Mondelli V, Cattaneo A, Murri MB, et al: Stress and inflammation reduce brain-derived neurotrophic factor expression in first-episode psychosis: a pathway to smaller hippocampal volume. J Clin Psychiatry 2011; 72:1677–1684
100.
Shivakumar V, Sreeraj VS, Subbanna M, et al: Differential impact of interleukin-6 promoter gene polymorphism on hippocampal volume in antipsychotic-naïve schizophrenia patients. Indian J Psychiatry 2020; 62:36–42
101.
Wu D, Lv P, Li F, et al: Association of peripheral cytokine levels with cerebral structural abnormalities in schizophrenia. Brain Res 2019; 1724:146463
102.
Prasad KM, Upton CH, Nimgaonkar VL, et al: Differential susceptibility of white matter tracts to inflammatory mediators in schizophrenia: an integrated DTI study. Schizophr Res 2015; 161:119–125
103.
Lesh TA, Careaga M, Rose DR, et al: Cytokine alterations in first-episode schizophrenia and bipolar disorder: relationships to brain structure and symptoms. J Neuroinflammation 2018; 15:165
104.
Fillman SG, Weickert TW, Lenroot RK, et al: Elevated peripheral cytokines characterize a subgroup of people with schizophrenia displaying poor verbal fluency and reduced Broca’s area volume. Mol Psychiatry 2016; 21:1090–1098
105.
Cannon TD, Chung Y, He G, et al: Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol Psychiatry 2015; 77:147–157
106.
Fillman SG, Cloonan N, Catts VS, et al: Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry 2013; 18:206–214
107.
Volk DW, Chitrapu A, Edelson JR, et al: Molecular mechanisms and timing of cortical immune activation in schizophrenia. Am J Psychiatry 2015; 172:1112–1121
108.
Lanz TA, Reinhart V, Sheehan MJ, et al: Postmortem transcriptional profiling reveals widespread increase in inflammation in schizophrenia: a comparison of prefrontal cortex, striatum, and hippocampus among matched tetrads of controls with subjects diagnosed with schizophrenia, bipolar or major depressive disorder. Transl Psychiatry 2019; 9:151
109.
Laskaris LE, Di Biase MA, Everall I, et al: Microglial activation and progressive brain changes in schizophrenia. Br J Pharmacol 2016; 173:666–680
110.
Najjar S, Pearlman DM: Neuroinflammation and white matter pathology in schizophrenia: systematic review. Schizophr Res 2015; 161:102–112
111.
van Kesteren CF, Gremmels H, de Witte LD, et al: Immune involvement in the pathogenesis of schizophrenia: a meta-analysis on postmortem brain studies. Transl Psychiatry 2017; 7:e1075
112.
Réus GZ, Fries GR, Stertz L, et al: The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015; 300:141–154
113.
Ching ASC, Kuhnast B, Damont A, et al: Current paradigm of the 18-kDa translocator protein (TSPO) as a molecular target for PET imaging in neuroinflammation and neurodegenerative diseases. Insights Imaging 2012; 3:111–119
114.
Plavén-Sigray P, Matheson GJ, Collste K, et al: Positron emission tomography studies of the glial cell marker translocator protein in patients with psychosis: a meta-analysis using individual participant data. Biol Psychiatry 2018; 84:433–442
115.
Marques TR, Ashok AH, Pillinger T, et al: Neuroinflammation in schizophrenia: meta-analysis of in vivo microglial imaging studies. Psychol Med 2019; 49:2186–2196
116.
Mondelli V, Ciufolini S, Belvederi Murri M, et al: Cortisol and inflammatory biomarkers predict poor treatment response in first episode psychosis. Schizophr Bull 2015; 41:1162–1170
117.
Nettis MA, Pergola G, Kolliakou A, et al: Metabolic-inflammatory status as predictor of clinical outcome at 1-year follow-up in patients with first episode psychosis. Psychoneuroendocrinology 2019; 99:145–153
118.
Mondelli V, Di Forti M, Morgan BP, et al: Baseline high levels of complement component 4 predict worse clinical outcome at 1-year follow-up in first-episode psychosis. Brain Behav Immun 2020; 88:913-9-15
119.
He X, Ma Q, Fan Y, et al: The role of cytokines in predicting the efficacy of acute stage treatment in patients with schizophrenia. Neuropsychiatr Dis Treat 2020; 16:191–199
120.
Miller BJ, Buckley P, Seabolt W, et al: Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry 2011; 70:663–671
121.
Grüber L, Bunse T, Weidinger E, et al: Adjunctive recombinant human interferon gamma-1b for treatment-resistant schizophrenia in 2 patients. J Clin Psychiatry 2014; 75:1266–1267
122.
Miller BJ, Dias JK, Lemos HP, et al: An open-label, pilot trial of adjunctive tocilizumab in schizophrenia. J Clin Psychiatry 2016; 77:275–276
123.
Girgis RR, Ciarleglio A, Choo T, et al: A randomized, double-blind, placebo-controlled clinical trial of tocilizumab, an interleukin-6 receptor antibody, for residual symptoms in schizophrenia. Neuropsychopharmacology 2018; 43:1317–1323
124.
Weickert T, Jacomb I, Lenroot R, et al: Reduction in peripheral C-reactive protein levels with canakinumab administration is related to reduced positive symptom severity in patients with schizophrenia and inflammation. Schizophr Bull 2019; 45:S318–S318
125.
Zalcman S, Savina I, Wise RA: Interleukin-6 increases sensitivity to the locomotor-stimulating effects of amphetamine in rats. Brain Res 1999; 847:276–283
126.
Plitman E, Iwata Y, Caravaggio F, et al: Kynurenic acid in schizophrenia: a systematic review and meta-analysis. Schizophr Bull 2017; 43:764–777
127.
Pillinger T, Osimo EF, Brugger S, et al: A meta-analysis of immune parameters, variability, and assessment of modal distribution in psychosis and test of the immune subgroup hypothesis. Schizophr Bull 2019; 45:1120–1133
128.
Smith SE, Li J, Garbett K, et al: Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 2007; 27:10695–10702
Information & Authors
Information
Published In


History
Published in print: Fall 2020
Published online: 6 November 2020
Keywords
Authors
Competing Interests
Dr. Miller has received research support from Acadia and Alkermes and honoraria from Psychiatric Times. Dr. Goldsmith reports no financial relationships with commercial interests.
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBMedia
Figures
FIGURE 1. Potential mechanisms underlying associations between inflammation and schizophreniaa
aMHC, major histocompatibility complex. bIDO, indoleamine 2,3-dioxygenase.
Other
Tables
References
References
1.
Purcell SM, Wray NR, Stone JL, et al: Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460:748–752
2.
Sekar A, Bialas AR, de Rivera H, et al: Schizophrenia risk from complex variation of complement component 4. Nature 2016; 530:177–183
3.
Shi J, Levinson DF, Duan J, et al: Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009; 460:753–757
4.
Brown AS, Derkits EJ: Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry 2010; 167:261–280
5.
Benros ME, Pedersen MG, Rasmussen H, et al: A nationwide study on the risk of autoimmune diseases in individuals with a personal or a family history of schizophrenia and related psychosis. Am J Psychiatry 2014; 171:218–226
6.
Nielsen PR, Benros ME, Mortensen PB: Hospital contacts with infection and risk of schizophrenia: a population-based cohort study with linkage of Danish national registers. Schizophr Bull 2014; 40:1526–1532
7.
Kirkpatrick B, Miller BJ: Inflammation and schizophrenia. Schizophr Bull 2013; 39:1174–1179
8.
Mazza MG, Lucchi S, Rossetti A, et al: Neutrophil-lymphocyte ratio, monocyte-lymphocyte ratio and platelet-lymphocyte ratio in non-affective psychosis: a meta-analysis and systematic review. World J Biol Psychiatry 2020; 21:326–338
9.
Miller BJ, Culpepper N, Rapaport MH: C-reactive protein levels in schizophrenia: a review and meta-analysis. Clin Schizophr Relat Psychoses 2014; 7:223–230
10.
Wang AK, Miller BJ: Meta-analysis of cerebrospinal fluid cytokine and tryptophan catabolite alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder, and depression. Schizophr Bull 2018; 44:75–83
11.
Chae J, Miller B: Beyond urinary tract infections (UTIs) and delirium: a systematic review of UTIs and neuropsychiatric disorders. J Psychiatr Pract 2015; 21:402–411
12.
Nitta M, Kishimoto T, Müller N, et al: Adjunctive use of nonsteroidal anti-inflammatory drugs for schizophrenia: a meta-analytic investigation of randomized controlled trials. Schizophr Bull 2013; 39:1230–1241
13.
Sommer IE, van Westrhenen R, Begemann MJ, et al: Efficacy of anti-inflammatory agents to improve symptoms in patients with schizophrenia: an update. Schizophr Bull 2014; 40:181–191
14.
Laan W, Grobbee DE, Selten JP, et al: Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2010; 71:520–527
15.
Müller N, Ulmschneider M, Scheppach C, et al: COX-2 inhibition as a treatment approach in schizophrenia: immunological considerations and clinical effects of celecoxib add-on therapy. Eur Arch Psychiatry Clin Neurosci 2004; 254:14–22
16.
Sommer IE, van Bekkum DW, Klein H, et al: Severe chronic psychosis after allogeneic SCT from a schizophrenic sibling. Bone Marrow Transplant 2015; 50:153–154
17.
Miyaoka T, Wake R, Hashioka S, et al: Remission of psychosis in treatment-resistant schizophrenia following bone marrow transplantation: a case report. Front Psychiatry 2017; 8:174
18.
Dickerson F, Severance E, Yolken R: The microbiome, immunity, and schizophrenia and bipolar disorder. Brain Behav Immun 2017; 62:46–52
19.
Ezeoke A, Mellor A, Buckley P, et al: A systematic, quantitative review of blood autoantibodies in schizophrenia. Schizophr Res 2013; 150:245–251
20.
Flatow J, Buckley P, Miller BJ: Meta-analysis of oxidative stress in schizophrenia. Biol Psychiatry 2013; 74:400–409
21.
Miller BJ, Goldsmith DR: Towards an immunophenotype of schizophrenia: progress, potential mechanisms, and future directions. Neuropsychopharmacology 2017; 42:299–317
22.
Woo JJ, Pouget JG, Zai CC, et al: The complement system in schizophrenia: where are we now and what’s next? Mol Psychiatry 2020; 25:114–130
23.
Netea MG, Domínguez-Andrés J, Barreiro LB, et al: Defining trained immunity and its role in health and disease. Nat Rev Immunol 2020; 20:375–388
24.
Adib-Conquy M, Cavaillon JM: Compensatory anti-inflammatory response syndrome. Thromb Haemost 2009; 101:36–47
25.
Miller AH, Raison CL: The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 2016; 16:22–34
26.
Louveau A, Herz J, Alme MN, et al: CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci 2018; 21:1380–1391
27.
Quan N, Banks WA: Brain-immune communication pathways. Brain Behav Immun 2007; 21:727–735
28.
Pollak TA, Drndarski S, Stone JM, et al: The blood-brain barrier in psychosis. Lancet Psychiatry 2018; 5:79–92
29.
D’Mello C, Le T, Swain MG: Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci 2009; 29:2089–2102
30.
Golofast B, Vales K: The connection between microbiome and schizophrenia. Neurosci Biobehav Rev 2020; 108:712–731
31.
Takeuchi O, Akira S: Pattern recognition receptors and inflammation. Cell 2010; 140:805–820
32.
Fleshner M: Stress-evoked sterile inflammation, danger associated molecular patterns (DAMPs), microbial associated molecular patterns (MAMPs) and the inflammasome. Brain Behav Immun 2013; 27:1–7
33.
Kim YK, Shin C: The microbiota-gut-brain axis in neuropsychiatric disorders: pathophysiological mechanisms and novel treatments. Curr Neuropharmacol 2018; 16:559–573
34.
Schwarz E, Maukonen J, Hyytiäinen T, et al: Analysis of microbiota in first episode psychosis identifies preliminary associations with symptom severity and treatment response. Schizophr Res 2018; 192:398–403
35.
Shen Y, Xu J, Li Z, et al: Analysis of gut microbiota diversity and auxiliary diagnosis as a biomarker in patients with schizophrenia: a cross-sectional study. Schizophr Res 2018; 197:470–477
36.
Yuan X, Zhang P, Wang Y, et al: Changes in metabolism and microbiota after 24-week risperidone treatment in drug naïve, normal weight patients with first episode schizophrenia. Schizophr Res 2018; 201:299–306
37.
Severance EG, Gressitt KL, Stallings CR, et al: Discordant patterns of bacterial translocation markers and implications for innate immune imbalances in schizophrenia. Schizophr Res 2013; 148:130–137
38.
Yolken RH, Severance EG, Sabunciyan S, et al: Metagenomic sequencing indicates that the oropharyngeal phageome of individuals with schizophrenia differs from that of controls. Schizophr Bull 2015; 41:1153–1161
39.
Gao L, Li Z, Chang S, et al: Association of interleukin-10 polymorphisms with schizophrenia: a meta-analysis. PLoS One 2014; 9:e90407
40.
Hudson ZD, Miller BJ: Meta-analysis of cytokine and chemokine genes in schizophrenia. Clin Schizophr Relat Psychoses 2018; 12:121–129B
41.
Shibuya M, Watanabe Y, Nunokawa A, et al: Interleukin 1 beta gene and risk of schizophrenia: detailed case-control and family-based studies and an updated meta-analysis. Hum Psychopharmacol 2014; 29:31–37
42.
Ligthart S, Vaez A, Võsa U, et al: Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. Am J Hum Genet 2018; 103:691–706
43.
Prins BP, Abbasi A, Wong A, et al: Investigating the causal relationship of c-reactive protein with 32 complex somatic and psychiatric outcomes: a large-scale cross-consortium Mendelian randomization study. PLoS Med 2016; 13:e1001976
44.
Gaughran F, O’Neill E, Sham P, et al: Soluble interleukin 2 receptor levels in families of people with schizophrenia. Schizophr Res 2002; 56:235–239
45.
Martínez-Gras I, García-Sánchez F, Guaza C, et al: Altered immune function in unaffected first-degree biological relatives of schizophrenia patients. Psychiatry Res 2012; 200:1022–1025
46.
Rapaport MH, Torrey EF, McAllister CG, et al: Increased serum soluble interleukin-2 receptors in schizophrenic monozygotic twins. Eur Arch Psychiatry Clin Neurosci 1993; 243:7–10
47.
Nunes SO, Matsuo T, Kaminami MS, et al: An autoimmune or an inflammatory process in patients with schizophrenia, schizoaffective disorder, and in their biological relatives. Schizophr Res 2006; 84:180–182
48.
Torrey EF, Bartko JJ, Lun ZR, et al: Antibodies to Toxoplasma gondii in patients with schizophrenia: a meta-analysis. Schizophr Bull 2007; 33:729–736
49.
Meyer U: Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry 2014; 75:307–315
50.
Zhang J, Luo W, Huang P, et al: Maternal C-reactive protein and cytokine levels during pregnancy and the risk of selected neuropsychiatric disorders in offspring: a systematic review and meta-analysis. J Psychiatr Res 2018; 105:86–94
51.
Allswede DM, Yolken RH, Buka SL, et al: Cytokine concentrations throughout pregnancy and risk for psychosis in adult offspring: a longitudinal case-control study. Lancet Psychiatry 2020; 7:254–261.
52.
Miller BJ, Culpepper N, Rapaport MH, et al: Prenatal inflammation and neurodevelopment in schizophrenia: a review of human studies. Prog Neuropsychopharmacol Biol Psychiatry 2013a; 42:92–100
53.
Cooper JD, Ozcan S, Gardner RM, et al: Schizophrenia-risk and urban birth are associated with proteomic changes in neonatal dried blood spots. Transl Psychiatry 2017; 7:1290
54.
Gardner RM, Dalman C, Wicks S, et al: Neonatal levels of acute phase proteins and later risk of non-affective psychosis. Transl Psychiatry 2013; 3:e228
55.
Blomström Å, Gardner RM, Dalman C, et al: Influence of maternal infections on neonatal acute phase proteins and their interaction in the development of non-affective psychosis. Transl Psychiatry 2015; 5:e502
56.
Khandaker GM, Pearson RM, Zammit S, et al: Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study. JAMA Psychiatry 2014; 71:1121–1128
57.
Khandaker GM, Zammit S, Burgess S, et al: Association between a functional interleukin 6 receptor genetic variant and risk of depression and psychosis in a population-based birth cohort. Brain Behav Immun 2018; 69:264–272
58.
Fusar-Poli P, Cappucciati M, Borgwardt S, et al: Heterogeneity of psychosis risk within individuals at clinical high risk: a meta-analytical stratification. JAMA Psychiatry 2016; 73:113–120
59.
Park S, Miller BJ: Meta-analysis of cytokine and C-reactive protein levels in high-risk psychosis. Schizophr Res 2019; doi:
60.
Stojanovic A, Martorell L, Montalvo I, et al: Increased serum interleukin-6 levels in early stages of psychosis: associations with at-risk mental states and the severity of psychotic symptoms. Psychoneuroendocrinology 2014; 41:23–32
61.
Goldsmith DR, Haroon E, Miller AH, et al: Association of baseline inflammatory markers and the development of negative symptoms in individuals at clinical high risk for psychosis. Brain Behav Immun 2019; 76:268–274
62.
Benros ME, Nielsen PR, Nordentoft M, et al: Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am J Psychiatry 2011; 168:1303–1310
63.
Eaton WW, Byrne M, Ewald H, et al: Association of schizophrenia and autoimmune diseases: linkage of Danish national registers. Am J Psychiatry 2006; 163:521–528
64.
Eaton WW, Pedersen MG, Nielsen PR, et al: Autoimmune diseases, bipolar disorder, and non-affective psychosis. Bipolar Disord 2010; 12:638–646
65.
Lachance LR, McKenzie K: Biomarkers of gluten sensitivity in patients with non-affective psychosis: a meta-analysis. Schizophr Res 2014; 152:521–527
66.
Liu S, Zhang X, Wang J, et al: Analysis of plasma autoantibodies for inflammatory cytokines in patients with first-episode schizophrenia among a Chinese population. J Neuroimmunol 2020; 341:577165
67.
Pollak TA, McCormack R, Peakman M, et al: Prevalence of anti-N-methyl-D-aspartate (NMDA) receptor [corrected] antibodies in patients with schizophrenia and related psychoses: a systematic review and meta-analysis. Psychol Med 2014; 44:2475–2487
68.
Goldsmith DR, Rapaport MH, Miller BJ: A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry 2016; 21:1696–1709
69.
Upthegrove R, Manzanares-Teson N, Barnes NM: Cytokine function in medication-naive first episode psychosis: a systematic review and meta-analysis. Schizophr Res 2014; 155:101–108
70.
Chen DC, Qi LY, Xiu MH, et al: Elevated serum levels of tumor necrosis factor-alpha in clozapine-associated obesity in chronic schizophrenia. Schizophr Res 2008; 106:367–368
71.
Haack M, Hinze-Selch D, Fenzel T, et al: Plasma levels of cytokines and soluble cytokine receptors in psychiatric patients upon hospital admission: effects of confounding factors and diagnosis. J Psychiatr Res 1999; 33:407–418
72.
Klemettilä JP, Kampman O, Seppälä N, et al: Cytokine and adipokine alterations in patients with schizophrenia treated with clozapine. Psychiatry Res 2014; 218:277–283
73.
O’Connell KE, Thakore J, Dev KK: Pro-inflammatory cytokine levels are raised in female schizophrenia patients treated with clozapine. Schizophr Res 2014; 156:1–8
74.
Schmitt A, Bertsch T, Tost H, et al: Increased serum interleukin-1beta and interleukin-6 in elderly, chronic schizophrenic patients on stable antipsychotic medication. Neuropsychiatr Dis Treat 2005; 1:171–177
75.
Kluge M, Schuld A, Schacht A, et al: Effects of clozapine and olanzapine on cytokine systems are closely linked to weight gain and drug-induced fever. Psychoneuroendocrinology 2009; 34:118–128
76.
Löffler S, Klimke A, Kronenwett R, et al: Clozapine mobilizes CD34+ hematopoietic stem and progenitor cells and increases plasma concentration of interleukin 6 in patients with schizophrenia. J Clin Psychopharmacol 2010; 30:591–595
77.
Maes M, Bosmans E, Kenis G, et al: In vivo immunomodulatory effects of clozapine in schizophrenia. Schizophr Res 1997; 26:221–225
78.
Pollmächer T, Hinze-Selch D, Mullington J: Effects of clozapine on plasma cytokine and soluble cytokine receptor levels. J Clin Psychopharmacol 1996; 16:403–409
79.
Lü LX, Guo SQ, Chen W, et al: Effect of clozapine and risperidone on serum cytokine levels in patients with first-episode paranoid schizophrenia. [in Chinese] J First Mil Med Univ 2004; 24:1251–1254
80.
Ajami A, Abedian F, Hamzeh Hosseini S, et al: Serum TNF-α, IL-10 and IL-2 in schizophrenic patients before and after treatment with risperidone and clozapine. Iran J Immunol 2014; 11:200–209
81.
Löffler S, Löffler-Ensgraber M, Fehsel K, et al: Clozapine therapy raises serum concentrations of high sensitive C-reactive protein in schizophrenic patients. Int Clin Psychopharmacol 2010; 25:101–106
82.
Maes M, Meltzer HY, Bosmans E: Immune-inflammatory markers in schizophrenia: comparison to normal controls and effects of clozapine. Acta Psychiatr Scand 1994; 89:346–351
83.
Monteleone P, Fabrazzo M, Tortorella A, et al: Plasma levels of interleukin-6 and tumor necrosis factor alpha in chronic schizophrenia: effects of clozapine treatment. Psychiatry Res 1997; 71:11–17
84.
Miller B, Essali N: Meta-analysis of cytokine levels and psychopathology in schizophrenia. Schizophr Bull 2019; 45:S191–S192
85.
Miller B, Mellor A, Buckley PF: Interleukin-6 and cognition in non-affective psychosis. Schizophr Bull 2013; 39:S242–S243
86.
Misiak B, Stańczykiewicz B, Kotowicz K, et al: Cytokines and C-reactive protein alterations with respect to cognitive impairment in schizophrenia and bipolar disorder: a systematic review. Schizophr Res 2018; 192:16–29
87.
Ribeiro-Santos R, de Campos-Carli SM, Ferretjans R, et al: The association of cognitive performance and IL-6 levels in schizophrenia is influenced by age and antipsychotic treatment. Nord J Psychiatry 2020; 74:187–193
88.
Lizano P, Lutz O, Ling G, et al: Association of choroid plexus enlargement with cognitive, inflammatory, and structural phenotypes across the psychosis spectrum. Am J Psychiatry 2019; 176:564–572
89.
Zhang L, Zheng H, Wu R, et al: The effect of minocycline on amelioration of cognitive deficits and pro-inflammatory cytokines levels in patients with schizophrenia. Schizophr Res 2019; 212:92–98
90.
Miller BJ, Graham KL, Bodenheimer CM, et al: A prevalence study of urinary tract infections in acute relapse of schizophrenia. J Clin Psychiatry 2013c; 74:271–277
91.
Laney D, Philip N, Miller BJ: Recurrent urinary tract infections in acute psychosis. Schizophr Res 2015; 164:275–276
92.
Monroe JM, Buckley PF, Miller BJ: Meta-analysis of anti-Toxoplasma gondii IgM antibodies in acute psychosis. Schizophr Bull 2015; 41:989–998
93.
Ahokas A, Rimón R, Koskiniemi M, et al: Viral antibodies and interferon in acute psychiatric disorders. J Clin Psychiatry 1987; 48:194–196
94.
Srikanth S, Ravi V, Poornima KS, et al: Viral antibodies in recent onset, nonorganic psychoses: correspondence with symptomatic severity. Biol Psychiatry 1994; 36:517–521
95.
Fellerhoff B, Laumbacher B, Mueller N, et al: Associations between Chlamydophila infections, schizophrenia and risk of HLA-A10. Mol Psychiatry 2007; 12:264–272
96.
Nudel R, Wang Y, Appadurai V, et al: A large-scale genomic investigation of susceptibility to infection and its association with mental disorders in the Danish population. Transl Psychiatry 2019; 9:283
97.
Miller BJ, Herzig KH, Jokelainen J, et al: Inflammation, hippocampal volume, and cognition in schizophrenia: results from the Northern Finland 1966 Birth Cohort. Eur Arch Psychiatry Clin Neuroscience 2020; doi:
98.
Kalmady SV, Venkatasubramanian G, Shivakumar V, et al: Relationship between interleukin-6 gene polymorphism and hippocampal volume in antipsychotic-naïve schizophrenia: evidence for differential susceptibility? PLoS One 2014; 9:e96021
99.
Mondelli V, Cattaneo A, Murri MB, et al: Stress and inflammation reduce brain-derived neurotrophic factor expression in first-episode psychosis: a pathway to smaller hippocampal volume. J Clin Psychiatry 2011; 72:1677–1684
100.
Shivakumar V, Sreeraj VS, Subbanna M, et al: Differential impact of interleukin-6 promoter gene polymorphism on hippocampal volume in antipsychotic-naïve schizophrenia patients. Indian J Psychiatry 2020; 62:36–42
101.
Wu D, Lv P, Li F, et al: Association of peripheral cytokine levels with cerebral structural abnormalities in schizophrenia. Brain Res 2019; 1724:146463
102.
Prasad KM, Upton CH, Nimgaonkar VL, et al: Differential susceptibility of white matter tracts to inflammatory mediators in schizophrenia: an integrated DTI study. Schizophr Res 2015; 161:119–125
103.
Lesh TA, Careaga M, Rose DR, et al: Cytokine alterations in first-episode schizophrenia and bipolar disorder: relationships to brain structure and symptoms. J Neuroinflammation 2018; 15:165
104.
Fillman SG, Weickert TW, Lenroot RK, et al: Elevated peripheral cytokines characterize a subgroup of people with schizophrenia displaying poor verbal fluency and reduced Broca’s area volume. Mol Psychiatry 2016; 21:1090–1098
105.
Cannon TD, Chung Y, He G, et al: Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol Psychiatry 2015; 77:147–157
106.
Fillman SG, Cloonan N, Catts VS, et al: Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry 2013; 18:206–214
107.
Volk DW, Chitrapu A, Edelson JR, et al: Molecular mechanisms and timing of cortical immune activation in schizophrenia. Am J Psychiatry 2015; 172:1112–1121
108.
Lanz TA, Reinhart V, Sheehan MJ, et al: Postmortem transcriptional profiling reveals widespread increase in inflammation in schizophrenia: a comparison of prefrontal cortex, striatum, and hippocampus among matched tetrads of controls with subjects diagnosed with schizophrenia, bipolar or major depressive disorder. Transl Psychiatry 2019; 9:151
109.
Laskaris LE, Di Biase MA, Everall I, et al: Microglial activation and progressive brain changes in schizophrenia. Br J Pharmacol 2016; 173:666–680
110.
Najjar S, Pearlman DM: Neuroinflammation and white matter pathology in schizophrenia: systematic review. Schizophr Res 2015; 161:102–112
111.
van Kesteren CF, Gremmels H, de Witte LD, et al: Immune involvement in the pathogenesis of schizophrenia: a meta-analysis on postmortem brain studies. Transl Psychiatry 2017; 7:e1075
112.
Réus GZ, Fries GR, Stertz L, et al: The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015; 300:141–154
113.
Ching ASC, Kuhnast B, Damont A, et al: Current paradigm of the 18-kDa translocator protein (TSPO) as a molecular target for PET imaging in neuroinflammation and neurodegenerative diseases. Insights Imaging 2012; 3:111–119
114.
Plavén-Sigray P, Matheson GJ, Collste K, et al: Positron emission tomography studies of the glial cell marker translocator protein in patients with psychosis: a meta-analysis using individual participant data. Biol Psychiatry 2018; 84:433–442
115.
Marques TR, Ashok AH, Pillinger T, et al: Neuroinflammation in schizophrenia: meta-analysis of in vivo microglial imaging studies. Psychol Med 2019; 49:2186–2196
116.
Mondelli V, Ciufolini S, Belvederi Murri M, et al: Cortisol and inflammatory biomarkers predict poor treatment response in first episode psychosis. Schizophr Bull 2015; 41:1162–1170
117.
Nettis MA, Pergola G, Kolliakou A, et al: Metabolic-inflammatory status as predictor of clinical outcome at 1-year follow-up in patients with first episode psychosis. Psychoneuroendocrinology 2019; 99:145–153
118.
Mondelli V, Di Forti M, Morgan BP, et al: Baseline high levels of complement component 4 predict worse clinical outcome at 1-year follow-up in first-episode psychosis. Brain Behav Immun 2020; 88:913-9-15
119.
He X, Ma Q, Fan Y, et al: The role of cytokines in predicting the efficacy of acute stage treatment in patients with schizophrenia. Neuropsychiatr Dis Treat 2020; 16:191–199
120.
Miller BJ, Buckley P, Seabolt W, et al: Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry 2011; 70:663–671
121.
Grüber L, Bunse T, Weidinger E, et al: Adjunctive recombinant human interferon gamma-1b for treatment-resistant schizophrenia in 2 patients. J Clin Psychiatry 2014; 75:1266–1267
122.
Miller BJ, Dias JK, Lemos HP, et al: An open-label, pilot trial of adjunctive tocilizumab in schizophrenia. J Clin Psychiatry 2016; 77:275–276
123.
Girgis RR, Ciarleglio A, Choo T, et al: A randomized, double-blind, placebo-controlled clinical trial of tocilizumab, an interleukin-6 receptor antibody, for residual symptoms in schizophrenia. Neuropsychopharmacology 2018; 43:1317–1323
124.
Weickert T, Jacomb I, Lenroot R, et al: Reduction in peripheral C-reactive protein levels with canakinumab administration is related to reduced positive symptom severity in patients with schizophrenia and inflammation. Schizophr Bull 2019; 45:S318–S318
125.
Zalcman S, Savina I, Wise RA: Interleukin-6 increases sensitivity to the locomotor-stimulating effects of amphetamine in rats. Brain Res 1999; 847:276–283
126.
Plitman E, Iwata Y, Caravaggio F, et al: Kynurenic acid in schizophrenia: a systematic review and meta-analysis. Schizophr Bull 2017; 43:764–777
127.
Pillinger T, Osimo EF, Brugger S, et al: A meta-analysis of immune parameters, variability, and assessment of modal distribution in psychosis and test of the immune subgroup hypothesis. Schizophr Bull 2019; 45:1120–1133
128.
Smith SE, Li J, Garbett K, et al: Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 2007; 27:10695–10702