VWMD clinically ranges from antenatal or early infantile onset with poor outcome (
2) to adult onset with slow progression, where the age at onset is a strong predictor of disease course (
3). Affected individuals typically have normal early development, followed by neurological deterioration triggered by stress-provoked episodes of rapid decline. Patients with adolescent or adult onset are more likely to exhibit cognitive problems at the time of or shortly after the disease onset. However, psychiatric disorders have been rarely reported as the only symptoms at the time of diagnosis (
3–
7) and usually are followed by neurologic manifestations within a few years (
6).
In women, VWMD can present as ovarioleukodystrophy, a condition in which leukodystrophy is associated with primary or secondary ovarian failure (
2,
3). While this could help in reaching a diagnosis in females, the diagnosis in adult males may be challenging, given the possible atypical onset and insidious clinical course.
EIF2B5 is by far the most commonly mutated gene among adults with VWMD, with the recurrent c.338G>A(p.Arg113His) mutation accounting for the majority of adult cases (
6), while c.260C>T(p.Ala87Val) is a founder
EIF2B3 VWMD mutation in the French Canadian population of Quebec (
8).
Here, we present the case of an adult male who presented with only psychiatric symptoms for more than 20 years before occurrence of the typical neurological symptoms and deterioration of VWMD.
Case Report
The patient came to our attention at the age of 41 for suspected genetic leukoencephalopathy. He was the second child of French Canadian consanguineous (first cousins) parents, originally from Matapedia, a small region in Quebec. His family history was remarkable for psychiatric illness on the maternal side: two of his mother’s brothers died by suicide in the context of drug addiction, and his grandmother had schizophrenia. There were no neurological disorders segregating in the family.
In terms of development, the patient had normal psychomotor and language acquisitions. During late childhood and adolescence, he started to exhibit severe behavioral problems, which retrospectively fit the criteria for disruptive behavior disorder: he would lose his temper quickly, he was frequently involved in violent fights with his peers, and he was defiant toward authority both at home and in school. In addition, he sexually abused a family member. He experienced his first episode of depression at age 16 when he attempted suicide by driving into the family house, causing considerable property damage. This led to his removal from the family home, and a residential treatment program was started. During this period, he did not receive a clear psychiatric diagnosis. His behavior disorder led to significant impairment in his social interactions and caused him to drop out of high school. He eventually worked as a truck driver until the age of 41 when he was finally diagnosed with VWMD.
Since age 16, the patient presented transient urinary incontinence that continued for many years without any other associated neurological symptoms. He experienced a second episode of depression at age 34 when he started to notice some difficulty concentrating as well as short-term memory impairment. For the depressive symptoms, he was prescribed venlafaxine without significant benefit. Four years later, he was admitted to the hospital for a suicide attempt in the context of a further episode of major depression. At that time, his family noticed, for the first time, mild gait instability, and the patient complained of occasional dizziness and lack of energy.
At age 40, the patient was involved in a truck accident, during which he lost consciousness and required hospital admission. Since the accident, he has experienced cognitive decline, progressive gait deterioration with recurrent falls, increasing fine motor impairment, and daily dizziness.
Neuroradiological Findings
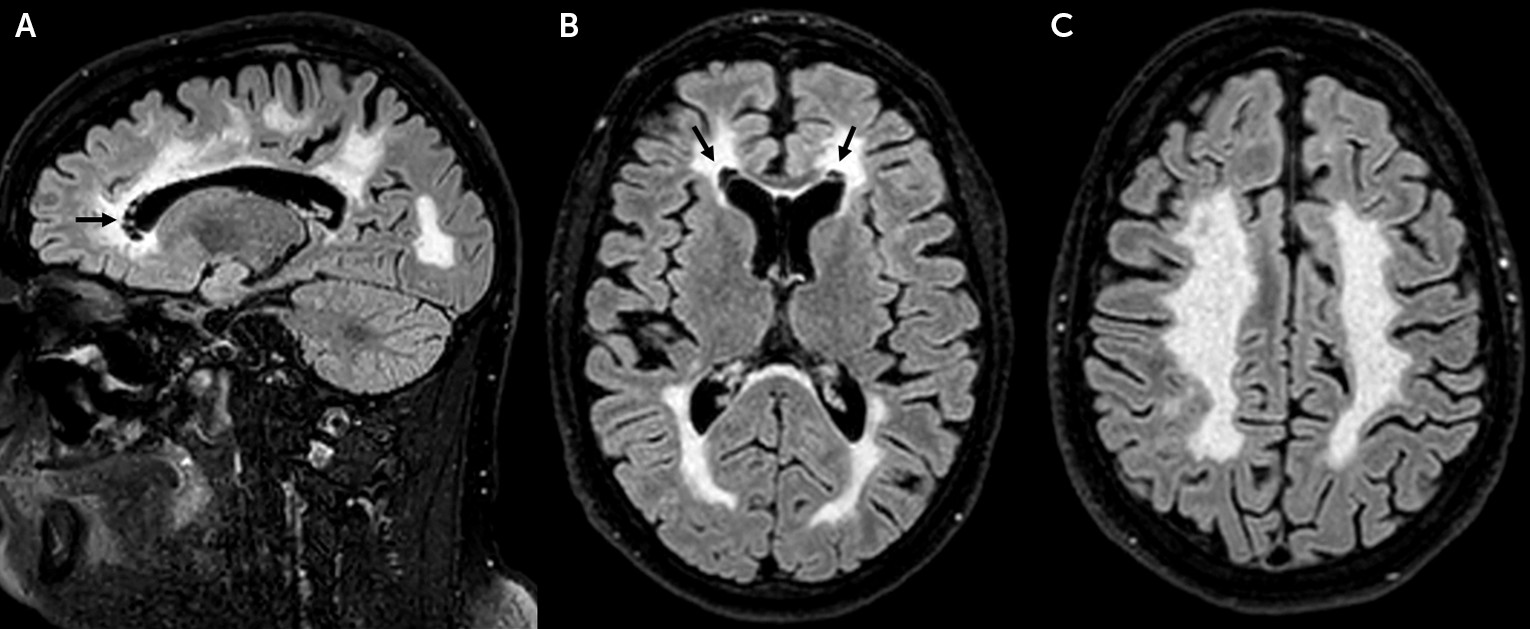
Following the truck accident, a head CT scan revealed the presence of extensive, symmetric white matter disease. Brain MRI (
Figure 1) confirmed the presence of bilateral, symmetric, and diffuse white matter abnormalities. Small areas of focal white matter rarefaction were noted bilaterally at the level of the frontal horns of the lateral ventricles. The U fibers, the external rim of the corpus callosum, the internal capsule, and cerebellar white matter were relatively spared.
Physical Examination
At the time of the patient’s clinical examination, he was still receiving monotherapy with venlafaxine. He was oriented and collaborative. He required help from his parents to recollect his personal and clinical history, he showed detachment from major events of his life (i.e., suicide attempts), and his mood appeared to be depressed. Neurological evaluation revealed mild increased tone, brisk reflexes in the lower limbs, difficulties with rapid alternating movements, and tandem gait. The patient’s gait was mildly ataxic. Given the constellation of the neuroradiological and clinical findings, VWMD was suspected.
Genetic Analysis
A gene panel for leukodystrophies, including the EIF2B1–5 genes, revealed the homozygous mutation c.260C>T(p.Ala87Val) in EIF2B3 (NM_020365.3).
Discussion
The long-standing psychiatric phenotype of our patient, in the absence of any neurological symptoms apart from sphincter dysfunction, represents a unique disease course never before reported, to our knowledge, among
EIF2B-related disorders. Psychiatric features and cognitive decline have been described as presenting symptoms in up to 24% of all VWMD patients and 11%−16% of adult-onset VWMD cases, but other neurological symptoms usually occur shortly afterward, within a few years (
2,
3,
6,
9). From our patient’s history, his gait and coordination were normal to the extent of working as a truck driver.
Isolated psychiatric symptoms can dominate the presentation of leukodystrophies in patients with onset in adolescence or adulthood (
10–
13). For this reason, we suggest that physicians take into account the possibility of genetic leukoencephalopathies in patients with long-standing psychiatric symptoms, especially if associated with nonspecific neurological symptoms (e.g., sphincter dysfunction). Interestingly, our patient presented with urinary incontinence since age 16, whereas in VWMD, this usually occurs during more advanced disease stages, when other neurological symptoms are already evident (
3). The predominant psychiatric presentation of our patient leads us to assume that VWMD may remain undiagnosed during the early stages of the disease, and its frequency may be underestimated among adult patients, especially when neurological symptoms are absent or nonspecific. Stress events, such as our patient’s truck accident, dramatically influence the course of the disease, leading to progressive motor decline.
The identification of a leukoencephalopathy was incidental in this patient, because it was only in the context of the truck accident that he underwent a CT scan, a test that started the neuroradiological assessment that led to the final diagnosis. The usefulness of brain MRI pattern recognition in genetic leukoencephalopathies is well known (
14). The neuroradiological phenotype of our patient is consistent with that of adult patients with VWMD in previous studies showing diffuse and symmetric white matter involvement with focal white matter rarefaction (
15). As demonstrated in a study of adult genetic leukoencephalopathies, the presence of white matter rarefaction should promptly suggest the diagnosis of VWMD (
7). The neuroradiological findings in our patient, in line with previous reports, confirm that anterior periventricular white matter rarefaction is a main distinctive neuroradiological feature in adults with VWMD.
EIF2B regulates protein synthesis rates under basal and cellular stress conditions, facilitating ternary complex formation and translation initiation (
1). Failure of the translation process of certain astrocytic mRNAs has been thought to be the underlying mechanism that induces degeneration of white matter through loss of astrocyte function, failure of astrocyte-microglia crosstalk, and secondary effect on both oligodendroglia and axons (
16). Several studies have shed light on the pathomechanism of
EIF2B, showing deregulation of endoplasmic reticulum function in astrocytes in mutant mice (
17) and prolonged state of translational hyperrepression in human VWMD cells that fail to recover from stress (
18). Although the pathomechanism underlying VWMD is becoming increasingly recognized, the reason for the broad phenotypic spectrum among
EIF2B-related disorders remains largely unknown.
Since the first report of the mutation more than a decade ago, c.260C>T(p.Ala87Val) has been rarely documented (
19–
22). Of note, it has been previously reported in two pediatric cases (
8) and in one adult female case (
15) in the Quebec population, showing variable disease course according to different age at disease onset. Our report confirms that c.260C>T(p.Ala87Val) in
EIF2B3 is a common mutation in the French Canadian population.
In conclusion, we have reported an isolated long-standing psychiatric presentation in an adult carrying an EIF2B3 mutation, broadening the clinical spectrum associated with eIF2B-related disorders. Importantly, we have highlighted the need for clinicians to consider a thorough neurological and neuroradiological assessment in individuals presenting with psychiatric features, especially when accompanied by mild nonspecific neurological symptoms, since the finding of focal white matter rarefaction on brain MRI would lead to a VWMD diagnosis.
Recognizing VWMD at an early stage is crucial in order to avoid stress-provoked episodes of rapid decline and may be critical for future therapeutic strategies.