Between 2012 and 2017, 18 patients with a history of substance abuse were evaluated at hospitals in Massachusetts for sudden-onset amnesia accompanied by bilateral hippocampal hyperintensities on diffusion-weighted MRI (
1–
3). In addition, two cases in West Virginia with this clinical radiological entity were recently reported (
4). Of these patients in Massachusetts and West Virginia, 19 had a history of opioid use or tested positive for opioids, including five toxicologically confirmed cases of fentanyl use (
2,
4). A spatiotemporal clustering of these reports parallels the rise of synthetic opioid use in the United States (
5). However, the full epidemiologic scope, mechanism of action, and predisposing factors underlying the syndrome remain unclear, as do the residual structural and functional sequelae of the hippocampal lesions. Here, we present the first case, to our knowledge, of this syndrome reported in the western United States in a patient with a history of opioid abuse who underwent detailed neuropsychological testing and serial brain MRIs before, during, and after presentation with amnestic symptoms.
Case Report
A 25-year-old right-handed man with a history of opioid abuse presented to a California emergency department with acute-onset anterograde amnesia following purported intravenous fentanyl use. The patient’s substance abuse history began during adolescence with infrequent recreational prescription opioids, then progressed to heroin and eventually to daily fentanyl use by his early 20s. His use of fentanyl was typically in combination with benzodiazepines and stimulants. At age 21, he developed a fear of losing his memory and obsessively wrote down detailed notes of intended activities and important conversations. Despite his fear of forgetting information, he held a 4.0 grade-point average in college. His heavy use of fentanyl continued until the age of 23, when he voluntarily entered a detoxification program and maintained sobriety for 18 months. Six days before drug relapse, he underwent an outpatient brain MRI. A high-resolution scan was ordered by the treating clinicians, presumably to rule out structural lesions given the patient’s persistent, subjective concern about his memory and reports of brief, stereotyped spells of a synthetic burning smell. Upon learning that his brain scan was unremarkable, the patient’s feeling of relief gave way to strong drug cravings. Later that same day, he reportedly purchased fentanyl and administered it intravenously, awakening hours later disoriented and amnestic. He repeatedly called a colleague, not remembering that they had just spoken. Concerned about the patient’s welfare, the colleague went to the patient’s home and found him disoriented, with needle marks on his arm, as well as two capped and used needles and a small bag of what appeared to be fentanyl. Dried emesis was found in the bathroom but with no evidence of blood. The patient was brought to a local emergency department approximately 36 hours after suspected opioid use.
Upon his arrival to the emergency department, the patient’s initial vital signs revealed mild sinus tachycardia, at 114 beats per minute, but were otherwise normal (temperature, 36.9°C, blood pressure, 124/84 mmHg, respiratory function, 18 respirations/minute with 98% O
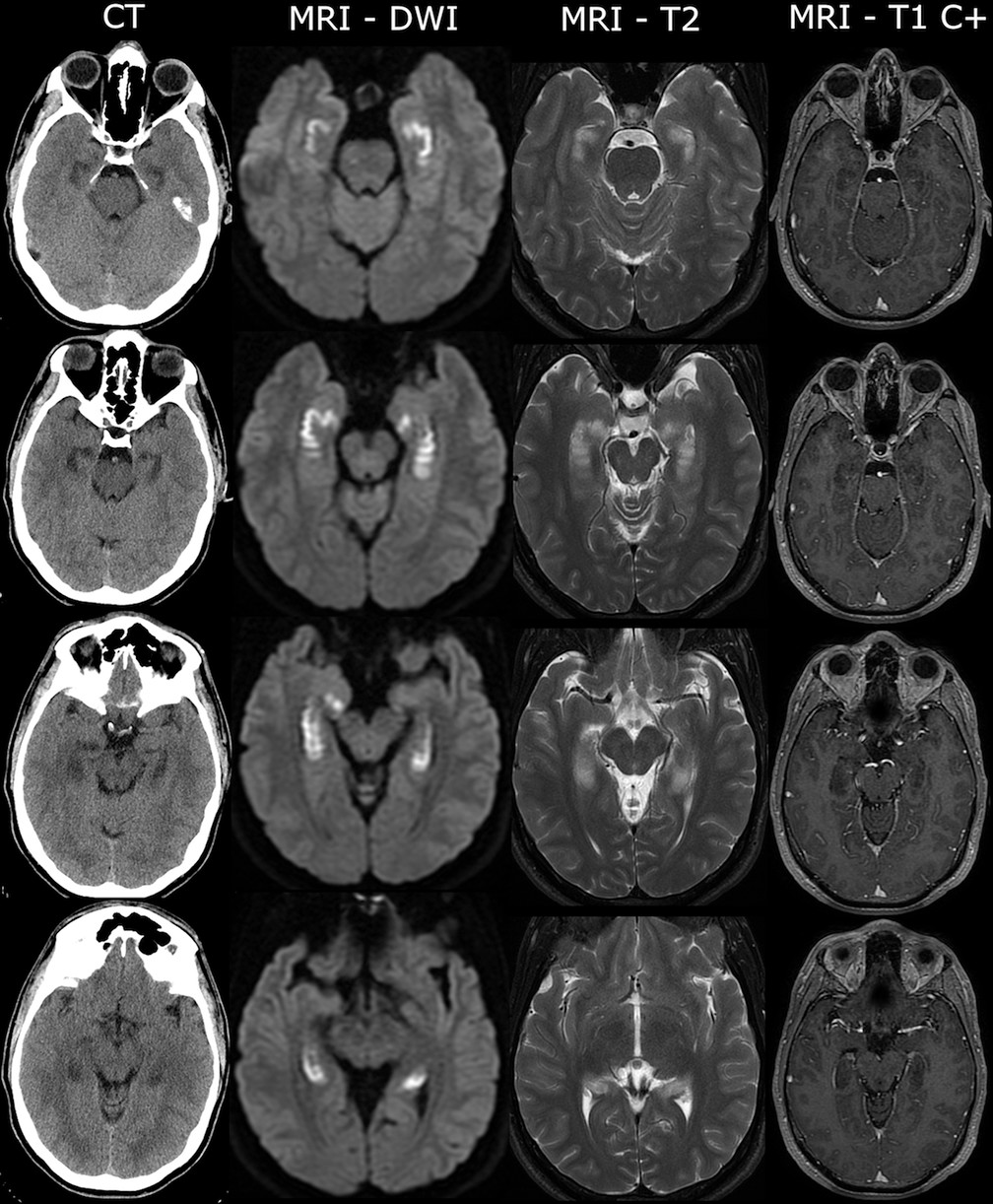
2 saturation on room air). On examination, the patient was distressed and repeated questions, asking “Did I relapse?” and “What city am I in?” His complete blood count and metabolic panel were notable for mild leukocytosis (11.2 K/μL) and elevation in liver enzymes (aspartate transaminase/alanine transaminase, 103 U/L/62 U/L), which normalized gradually over 3 days. A urine and serum toxicology screen was negative for alcohol, amphetamines, barbiturates, benzodiazepines, cocaine, methadone, opioids, and cannabinoids. A noncontrast head computerized tomography (CT) scan revealed hypoattenuation of the brain parenchyma in mesial temporal structures bilaterally (
Figure 1). Initial treatment included intravenous fluids, thiamine, and acyclovir due to concerns about viral encephalitis. Basic CSF examinations were normal (glucose level, 56 mg/dL, protein level, 40 mg/dL, <5 cells per mm
3), and polymerase chain reactions for herpes simplex virus nucleic acids type 1 and 2 were negative.
Brain MRI revealed bilateral restricted diffusion of the hippocampi without abnormal gadolinium enhancement (
Figure 1). MR angiography scans of the head and neck were normal. No epileptiform activity was identified on 48-hour continuous EEG. Erythrocyte sedimentation rate and C-reactive protein levels were elevated at 40 mm/hour (normal, <15) and 52.2 mg/L (normal, <8.0), respectively. Further metabolic, infectious, and autoimmune screenings (B
12, thyroid-stimulating hormone, antinuclear antibody, proteinase 3 and myeloperoxidase antibodies, HIV, syphilis, hepatitis A, B, and C, and CSF Gram stain and culture) were unremarkable. Neoplastic work-up included a contrast CT scan of the neck, chest, abdomen, and pelvis, which was unremarkable. Brief bedside cognitive evaluations revealed poor verbal recall and variable recall of recent autobiographical details with relative preservation of remote autobiographical memory.
Four months later, the patient came to the University of California at San Francisco Memory and Aging Center for research evaluation. His general neurological examination was normal. Results from neuropsychological testing revealed declarative memory deficits with severe episodic and mild semantic memory changes (for further details, see Table S1 in the online supplement). Specifically, the patient consistently performed under the 5th percentile compared with age- and education-matched control subjects on tests of delayed recall 10–20 minutes after exposure to various stimuli, which included word lists, stories, odors, and visual forms. The patient’s discrimination of recently learned information was poor, despite cueing and recognition. Below-average performance (range, 6th–37th percentile) was noted on language tasks, such as confrontational naming and semantic fluency.
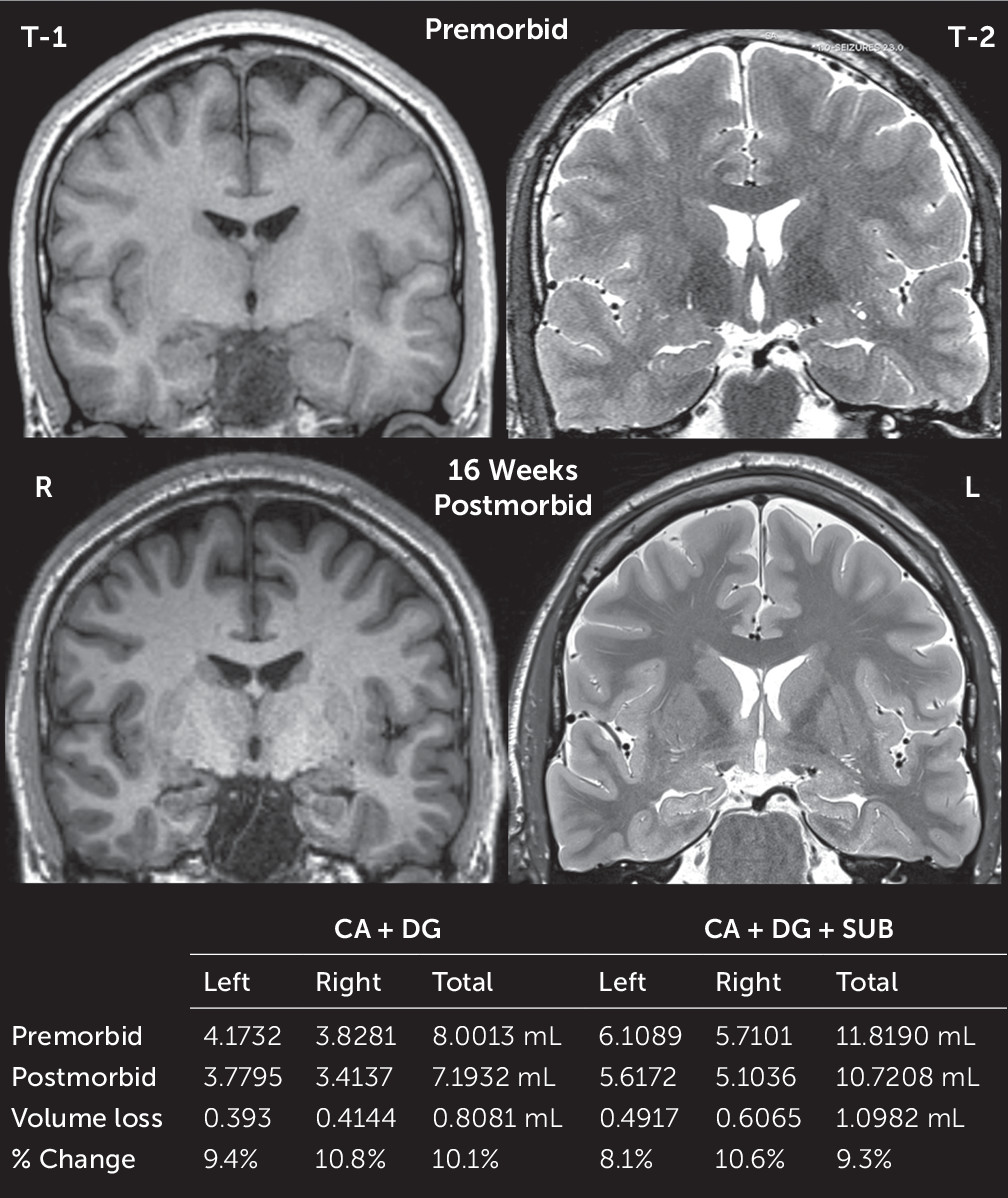
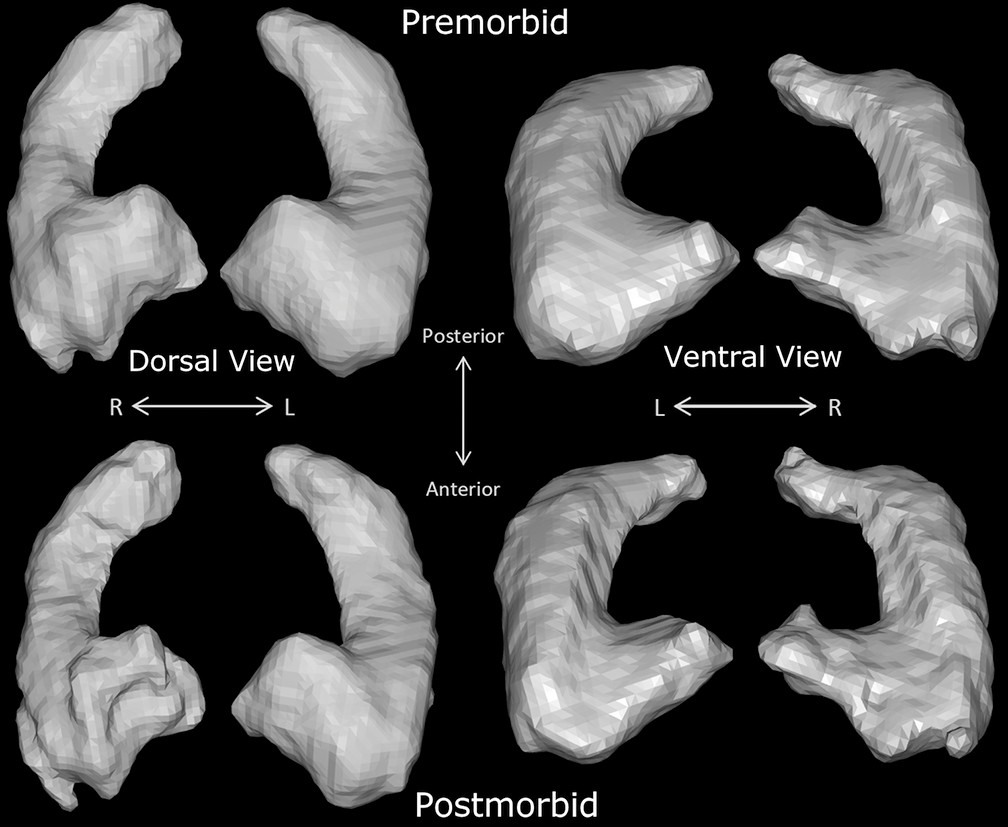
Comparison of pre- and postmorbid high-resolution brain MRI scans revealed approximately 10% volume loss in the cornu ammonis (CA), dentate, and subiculum hippocampal subfields (
Figures 2 and
3) (for further details, see the
online supplement). Diffusion-weighted MRI signals appeared normalized on postmorbid imaging. To investigate possible patient-specific predisposing factors, serum autoimmune antibody screening was conducted and revealed negative findings for
N-methyl-
d-aspartate, α-amino-3-hydroxy-5-methylisoxazole-4-propionate, GABA-A, GABA-B, metabotropic glutamate receptor 1 (mGluR1) and mGluR5, leucine rich glioma inactivated 1, contactin-associated protein-like 2, dipeptidyl-peptidase-like protein-6,
Neurexin3, and
IgLON5, which did not show any staining on rodent brain slices.
Discussion
This is the first case, to our knowledge, of an opioid-related amnestic syndrome identified in the western United States. Similar to reported cases from the eastern United States (
6), acute neuroimaging demonstrated gyriform signal changes in the hippocampi. We sought to understand the structural and functional sequelae of these lesions through neuropsychological assessment and quantitative imaging comparisons of the hippocampal formation. Pre- and postmorbid high-resolution scans revealed longitudinal structural change in the hippocampi 4 months after the patient’s acute neuronal injury. An approximate 10% structural hippocampal gray matter loss accompanied functional decline in cognition, evidenced by severe deficits in delayed episodic memory and mild deficits in semantic fluency.
In the above case, interpretation of the cause of the amnestic syndrome was limited by the drugs tested for in routine urine and serum immunoassays, which typically do not include fentanyl or other “designer” opioids. Unidentified synthetic drugs, adulterants, or contaminants cannot be ruled out as contributory in this case. The patient’s long history of fentanyl abuse and confirmatory toxicology from similar cases strengthen an association between fentanyl and this amnestic syndrome and underscore the importance of pursuing confirmatory toxicology in patients with similar presentations (
2,
6).
The hippocampal formation is susceptible to injury from a number of causes, such as hypoxemia, ischemia, hypoglycemia, seizures, limbic encephalitis, and paraneoplastic and nonparaneoplastic autoimmune-mediated limbic encephalopathies (
7). The majority of these potential causes were unlikely in the above case given that examination of infectious disease (e.g., CSF herpes simplex virus 1 and 2 polymerase chain reaction, serum rapid plasma reagin, and HIV) was unrevealing, continuous EEG was without epileptiform activity, an autoimmune encephalitis panel was negative, and CT body imaging did not reveal an occult neoplasm. Our patient was normoglycemic on presentation and lacked a history of diabetes. In addition, obstructive ischemia was an unlikely mechanism, given the bilateral and symmetric pattern of gyriform diffusion-weighted MRI abnormalities, which did not conform to typical vascular irrigation patterns. Hypoxic injury can result from hypoxemia, anemia, ischemia, or histotoxic insults. Anemic, ischemic, and histotoxic etiologies were deemed less likely given the lack of evidence for cardiac arrest, low suspicion for poisoning (e.g., cyanide and carbon monoxide), and normal serum hemoglobin, mean corpuscular volume, and B
12 levels.
Hypoxemic hypoxia remains a possible contributing factor. Global hypoventilation due to opioid-induced respiratory depression is well documented. Hypoventilation is mediated centrally by μ- and κ-opioid receptor binding in the ventrolateral medulla, which conducts efferent impulses along C4 and C5 ventral roots controlling respiratory frequency and inspiratory time (
8,
9). Acute global hypoxic-ischemic or hypoxemic-hypoxic episodes lead to a pattern of nerve cell damage in vulnerable neuronal populations, including pyramidal cells of Sommer’s sector of the hippocampus (e.g., CA1 > CA3), cerebellar Purkinje cells, small- and medium-sized striatal neurons, and neocortical layers III, V, and VI (
10–
12). Mechanistically, hypoxemia as a result of opioid-induced respiratory suppression might explain selective lesions for CA1 Sommer sector neurons, but our quantified subfield analysis suggests neuronal loss in hippocampal formation subfields other than CA1. Given that the patient’s total cerebral gray matter volumes pre- and postmorbid differed nominally by approximately 1%, extrahippocampal neurons susceptible to hypoxemia retained their integrity. If hypoxemia were the only mechanism of hippocampal injury, then one might expect many more prospective patients with this amnestic syndrome, given that in the United States alone, from 1996 to 2014, 26,500 individuals with opioid overdoses were administered naloxone (
13); presumably, variable periods of hypoventilation or cardiopulmonary arrest necessitated use of this reversal agent in a significant number of these cases.
A hypoxemic-hypoxic state taken together with patient-specific, contextual, or direct toxin-related effects on the hippocampus might explain these emerging cases of opioid-related amnesia. Variable genetic expression patterns in the hippocampus influence key pathways related to cerebral blood flow, calcium signaling, and apoptosis (
14,
15). Direct neurotoxic effects of opioids have been shown, including formation of reactive oxygen species and oxidative cell injury, reduced glutathione levels, and induction of apoptosis (
16). Evidence of deleterious acute and chronic opioid-mediated effects on hippocampal neuronal structure and function have been shown in animal and human studies (
6,
17). In relation to the case history of the patient presented above, one may wonder whether chronic opioid effects on hippocampal neurophysiology preconditioned for excitotoxic injury in the setting of transient hypoxia related to reintroduction of the offending agent following prolonged sobriety.
Fentanyl and its analogs are high-potency μ-opioid receptor agonists (
18). μ-opioid receptors are present in varying densities in all hippocampal subfields (
19). The predominant sites of μ-receptor expression in the hippocampal formation are GABA-ergic interneurons, which modulate principal cell excitation. μ-Opioid G-receptor signal transduction modulates potassium and calcium ion channel currents causing cellular hyperpolarization and inhibits tonic neural activity (
20). Within the hippocampus, the GABAergic tonic inhibitory signal modulates downstream excitability. Hypoxic conditions also lead to CA1 neuronal hyperpolarization (as a result of activation in K
ATP channels [
21]) and induce spreading depression starting in CA1 or CA3 (
22). However, in the setting of toxic-metabolic abnormalities, spreading depression propagates from CA1 into other subfields and independently arises from the subiculum or dentate (
23). Rat models suggest that exogenous opiate exposure exaggerates CA1 neuronal hypoxic injury and threatens neuronal integrity across the hippocampal formation (
24).
The neuropsychological data also point toward a more extensive hippocampal injury beyond the CA1 region. In the above case, recognition and discrimination of newly learned information was very poor, and the patient demonstrated at least mild deficits in semantic memory (e.g., semantic fluency <10th percentile), which suggests more widely distributed damage to the hippocampal-dependent declarative memory system, including CA4/CA3 pyramidal cells and dentate gyrus granule cells (
25). Understanding the context and evolution of opioid-related effects on hippocampal structure and function might provide insights into other conditions that affect episodic and semantic memory.
In this case of opioid-related acute amnestic syndrome in the western United States, pre- and postmorbid imaging comparisons revealed significant hippocampal gray matter volume loss, which accompanied clinical neuropsychological deficits in episodic memory and semantic function. This lesion-correlational approach underscores the function of hippocampal subfields for efficient episodic and semantic information processing, maintenance, recall, and recognition. Given the scope of the opioid epidemic in the United States, physicians should be aware of this focal clinical-radiological entity.