Clinical Features, Immunotherapy, and Outcomes of Anti-Leucine-Rich Glioma-Inactivated-1 Encephalitis
Abstract
Objective:
Methods:
Results:
Conclusions:
Methods
Study Participants
Data Collection
Treatment and Prognostic Evaluation
Statistical Approach
Results
Clinical Features
| Characteristic | N | Median | Total N | % | IQR |
|---|---|---|---|---|---|
| Age (years) | 57 | 43 | 44–65 | ||
| Male | 28 | 43 | 65.1 | ||
| Duration (days) | 60 | — | 37.0–127.0 | ||
| Initial reported symptoms | — | ||||
| Seizures | 18 | 43 | 41.9 | ||
| Memory impairment | 13 | 43 | 30.2 | ||
| Neuropsychiatric symptoms | 10 | 43 | 23.3 | ||
| Sleep disturbances | 1 | 43 | 2.3 | ||
| Movement abnormalities | 1 | 43 | 2.3 | ||
| Main clinical features | — | ||||
| Fever | 7 | 43 | 16.3 | ||
| Seizure | 43 | 43 | 100.0 | ||
| Status epilepticus | 10 | 43 | 23.3 | ||
| Faciobrachial dystonic seizures | 19 | 43 | 44.2 | ||
| Cognitive dysfunction | 41 | 43 | 95.3 | ||
| Neuropsychiatric symptoms | 33 | 43 | 76.7 | ||
| Behavioral dyscontrol | 18 | 33 | 54.5 | ||
| Visual and auditory hallucination | 17 | 33 | 51.5 | ||
| Nonsensical speech | 16 | 33 | 48.5 | ||
| Personality changes | 8 | 33 | 24.4 | ||
| Irritability | 6 | 33 | 18.2 | ||
| Anxiety | 5 | 33 | 15.2 | ||
| Sleep disturbances | 25 | 43 | 58.1 | ||
| Disturbance of consciousness | 21 | 43 | 48.8 | ||
| Movement abnormalities | 16 | 43 | 37.2 | ||
| Autonomic dysfunction | 14 | 43 | 32.6 | ||
| Serum sodium (mmol/L) | 133 | 43 | 130–138 | ||
| Hyponatremia (<135 mmol) | 22 | 43 | 51.2 | ||
| Increased pressure | 8 | 43 | 18.6 | ||
| CSF pleocytosis (>8 leukocytes/mL) | 7 | 43 | 16.3 | ||
| Increased CSF protein (>45 mg/dL) | 9 | 43 | 20.9 | ||
| Intrathecal synthesis of oligoclonal bands | 8 | 43 | 18.6 | ||
| LGI1 Abs positive in serum and CSF at admission | 43 | 43 | 100.0 | ||
| LGI1 Abs positive in CSF or serum at follow-up | 4 | 22 | 18.2 | ||
| LGI1 Abs negative at follow-up | 18 | 22 | 81.8 | ||
| Abnormal EEG at admission | 31 | 43 | 72.1 | ||
| Focal slow activity | 10 | 31 | 32.3 | ||
| Diffuse slow activity | 8 | 31 | 25.8 | ||
| Epileptic activity | 13 | 31 | 41.9 | ||
| Normal | 12 | 27.9 | |||
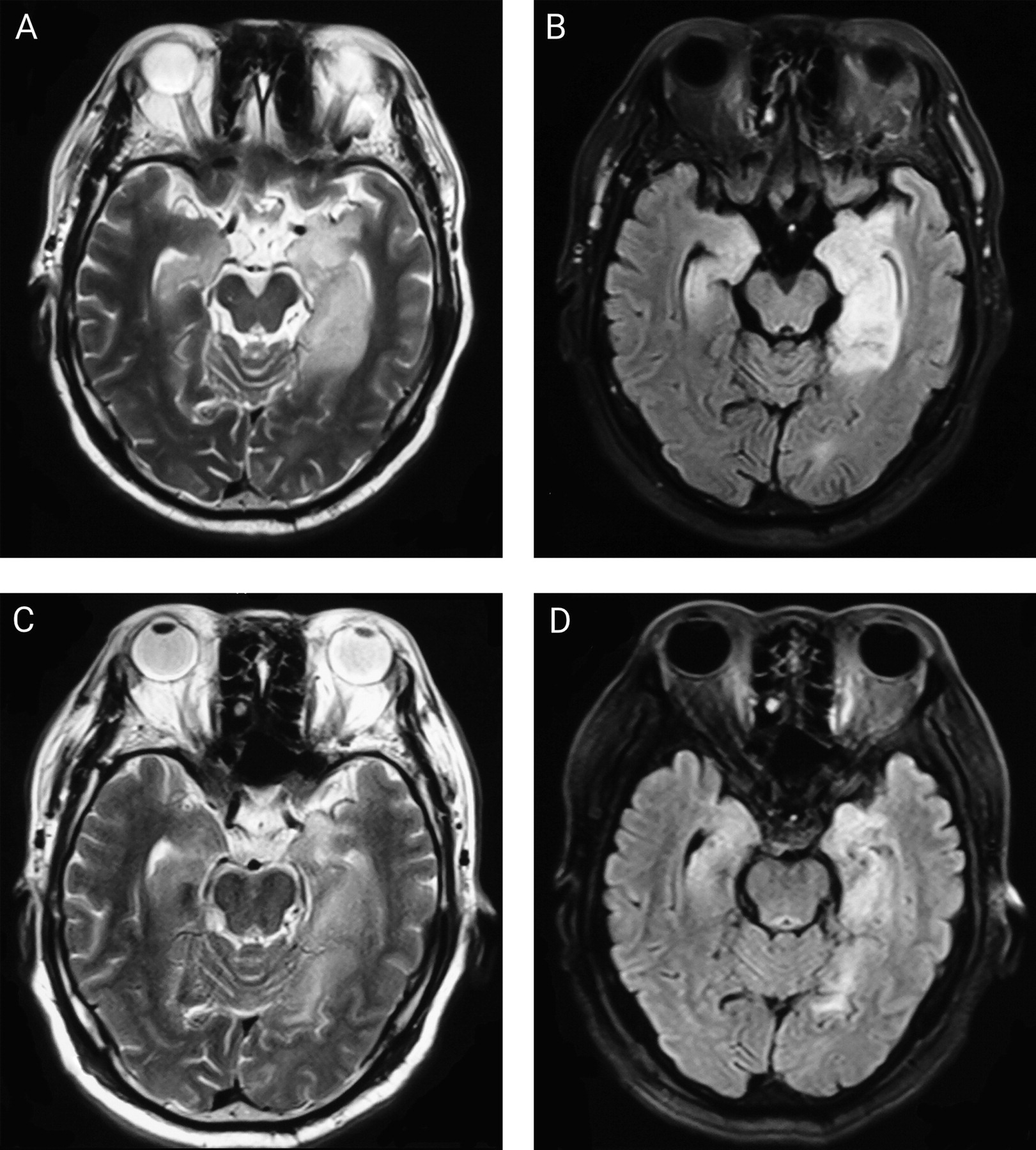
| Abnormal MRI at presentation | 30 | 43 | 69.8 | ||
| Hippocampal | 23 | 30 | 76.7 | ||
| Temporal lobe | 12 | 30 | 40.0 | ||
| Basal ganglia | 6 | 30 | 20.0 | ||
| Insular cortex | 3 | 30 | 10.0 | ||
| Other lesions | 9 | 30 | 30.0 | ||
| Normal MRI | 13 | 43 | 30.2 | ||
| MRI at follow-up | 34 | 43 | 79.1 | ||
| Hippocampal or mesial temporal atrophy | 20 | 34 | 58.8 | ||
| Hippocampal T2 hyperintensity | 6 | 34 | 17.6 | ||
| Normal | 5 | 34 | 14.7 | ||
| Local hypoperfusion on brain SPECT | 8 | 10 | 0.8 | ||
| Time to initiate immunotherapy (days) | 64 | — | 43.0–127.0 | ||
| Corticosteroids plus IVIg | 21 | 43 | 48.8 | ||
| Corticosteroids or IVIg | 22 | 43 | 30.2 | ||
| Mycophenolate mofetil | 4 | 43 | 9.3 | ||
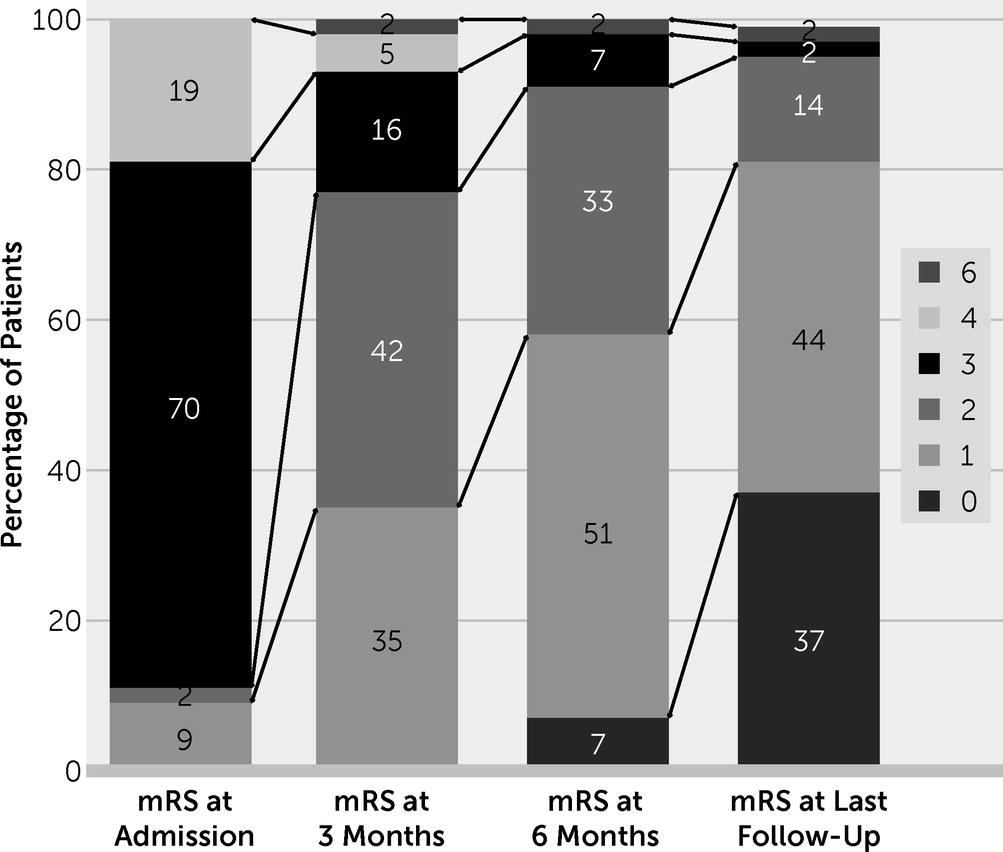
| mRS score at admission | 3 | — | 3.3–4.0 | ||
| mRS score at last follow-up | 1 | — | 0–1 | ||
| ≤2 | 41 | 43 | 95.3 | ||
| 3–5 | 1 | 43 | 2.3 | ||
| 0–1 | 15 | 43 | 34.9 | ||
| Deceased at last follow-up | 1 | 43 | 2.3 | ||
| Relapse | 10 | 43 | 23.3 | ||
| Length of hospitalization (days) | 16 | — | 13.0–22.0 | ||
| Period of follow-up (months) | 21.5 | — | 7.0–43.0 |

Auxiliary Investigations
Treatment
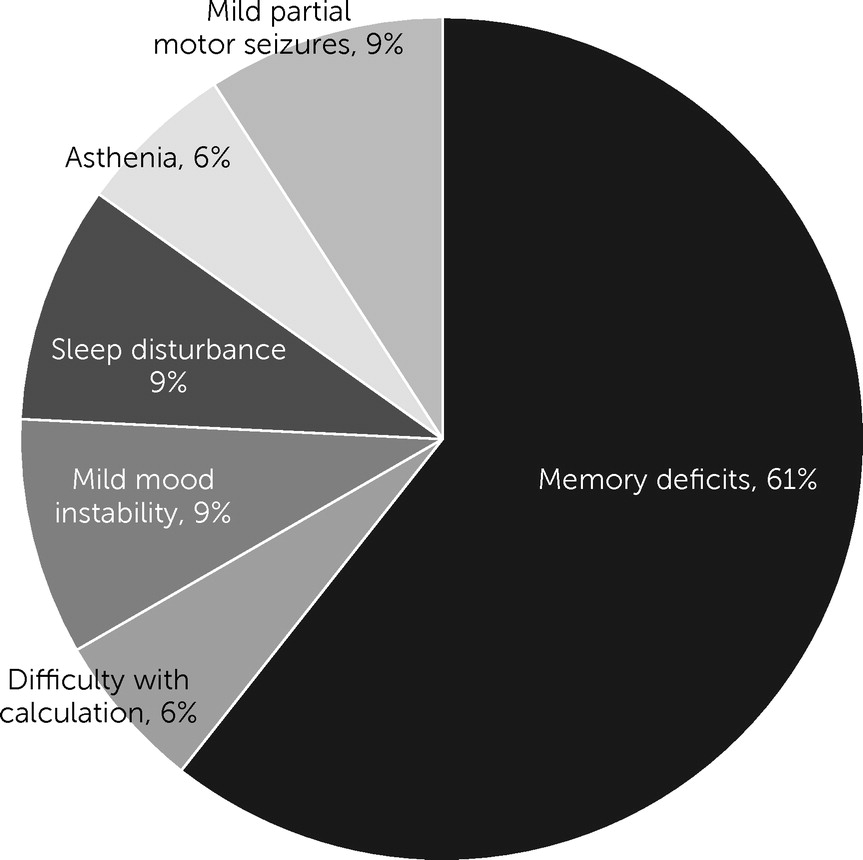
Outcomes and Analysis of Factors
| Functionally independent group (N=25) | Disabled group (N=18) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Variable | N | Median | % | IQR | N | Median | % | IQR | p |
| Age (years) | 58.0 | 44.0–64.5 | 56.0 | 45.5–65.5 | 0.892 | ||||
| Male | 15 | 60.0 | 9 | 50.0 | 0.550 | ||||
| Duration | 60.0 | 38.5–105.0 | 60.0 | 36.0–187.5 | 0.585 | ||||
| Type of onset (<3 months) | 16 | 64.0 | 11 | 61.1 | 1.000 | ||||
| Fever | 3 | 12.0 | 4 | 22.2 | 0.427 | ||||
| Seizure | 21 | 84.0 | 17 | 94.4 | 0.380 | ||||
| Faciobrachial dystonic seizures | 13 | 52.0 | 6 | 33.3 | 0.351 | ||||
| Cognitive dysfunction | 23 | 92.0 | 18 | 100.0 | 0.502 | ||||
| Neuropsychiatric symptoms | 20 | 80.0 | 13 | 72.2 | 0.717 | ||||
| Disturbances of consciousness | 11 | 44.0 | 10 | 55.5 | 0.543 | ||||
| Sleep disturbances | 14 | 56.0 | 11 | 61.1 | 0.765 | ||||
| Movement | 10 | 40.0 | 6 | 33.3 | 0.755 | ||||
| Autonomic dysfunction | 8 | 32.0 | 7 | 38.9 | 0.750 | ||||
| Serum sodium (mmol/L) | 137 | 131.0–139.0 | 130 | 126.0–136.5 | 0.018b | ||||
| Hyponatremia (<145 mmol/L) | 11 | 44.0 | 11 | 61.1 | 0.358 | ||||
| CSF pleocytosis (>8 cells/dl) | 7 | 28.0 | 5 | 27.8 | 1.000 | ||||
| Protein level (mg/dl) | 30.0 | 24.0–41.5 | 32.5 | 18.8–48.5 | 0.455 | ||||
| Abnormal EEG | 12 | 48.0 | 15 | 83.3 | 0.026* | ||||
| Abnormal MRI | 15 | 60.0 | 13 | 72.2 | 0.523 | ||||
| Involvement of limbic system | 18 | 72.0 | 11 | 61.1 | 0.335 | ||||
| Hippocampal atrophy | 14 | 56.0 | 9 | 50.0 | 0.763 | ||||
| Steroids plus IVIg | 15 | 60.0 | 10 | 55.6 | 1.000 | ||||
| Steroids or IVIg | 10 | 40.0 | 8 | 44.4 | 1.000 | ||||
| Immunosuppressant | 2 | 8.0 | 2 | 11.1 | 1.000 | ||||
| Time to start treat (<1 month) | 5 | 20.0 | 3 | 16.7 | 1.000 | ||||
| mRS score on admission | 2 | 1–4 | 2 | 0–4 | 0.006* | ||||
| Relapse | 6 | 24.0 | 4 | 22.2 | 0.594 | ||||
| Disability index | Odds ratio | 95% CI | p |
|---|---|---|---|
| Age (years) | 1.012 | 0.941, 1.088 | 0.749 |
| Sex | 1.291 | 0.411, 5.420 | 0.543 |
| Type of onset (subacute <3 months) | 0.166 | 0.009, 3.170 | 0.233 |
| Duration | 0.100 | 0.996, 1.012 | 0.291 |
| Serum sodium (mmol/L) | 0.806 | 0.663, 0.981 | 0.031 |
| Protein level of CSF (mg/dl) | 0.064 | 0.03, 1.641 | 0.097 |
| EEG abnormality | 0.109 | 0.011, 1.104 | 0.061 |
| MRI abnormality | 0.313 | 0.046, 2.143 | 0.237 |
| Involvement of limbic system | 2.307 | 0.47, 11.2 | 0.310 |
| Therapy with steroids plus IVIg | 1.678 | 0.230, 12.235 | 0.610 |
| Time to treatment | 0.271 | 0.015, 4.887 | 0.376 |
| mRS score on admission | 5.398 | 1.006, 28.970 | 0.050 |
Discussion
Conclusions
Footnote
References
Information & Authors
Information
Published In


History
Keywords
Authors
Funding Information
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBLogin options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).