Case Presentation
A 74-year-old man presented for evaluation of swallowing difficulties, weakness, and memory concerns. The patient had been in his usual state of health until approximately 10 years prior to presentation, when at age 64, he first experienced difficulty swallowing. He underwent two dilatation procedures for a suspected esophageal stricture that failed to ameliorate the problem. Although he transitioned to eating a mechanical soft diet at home, he experienced coughing while eating and took several hours to finish a meal. He unintentionally lost approximately 30 pounds over the course of 2 years. Five years before presentation, he developed slowly progressive weakness in his hands and proximal legs, with notable difficulties standing from a seated position and ascending and descending stairs. Four years prior to this evaluation, he fell down the stairs at a train station, sustaining a small subarachnoid hemorrhage in the left posterior sylvian fissure and a cortical contusion within the right anterior temporal lobe. Over the prior 2 years, he had lost the ability to write due to finger weakness, required self-fashioned tools to open jars, transitioned to walking with a cane, and fell again at home when trying to load paper into his printer.
Over this same 2-year period, he experienced cognitive slowing, distractibility, difficulties retrieving words, and difficulties with memory of recent events and information. Others had noted that the patient would lose track of what exercises he had been doing when between sets at the gym and would repeat questions and statements more frequently than he had previously. As a recently retired professor of computer science, the patient had been engaged in research and supervising students until shortly before his evaluation. Cognitive changes had not contributed to his decision to retire or prevented him from engaging in any of his usual activities. When he and his wife applied for entry into a retirement community, his score on a cognitive screening test prompted referral for further assessment.
He had no history of somatosensory symptoms, pain, autonomic symptoms, shortness of breath, neuropsychiatric symptoms (including those involving mood, anxiety, psychosis, and personality changes), or sleep-related issues except for severe obstructive sleep apnea for which he was adherent with continuous positive airway pressure (CPAP) treatment. His medical and surgical histories also included hypertension, dyslipidemia, coronary artery disease, prostate cancer treated with prostatectomy, sensorineural hearing loss treated with hearing aids, cataracts, and right hip osteoarthritis treated with total hip replacement. His medications included aspirin, atenolol, and atorvastatin. His family history included a father who developed memory difficulties at age 74 and died the following year, but there was no family history of additional neurological illness, including neuromuscular disorders.
Questions: What are general diagnostic considerations regarding the motor and cognitive symptoms? How might a neurological examination help to refine the list of possible diagnoses?
An important consideration from the start is whether a single underlying condition accounts for both the cognitive and motor symptoms or whether separate conditions are responsible. The sequence of symptom development in which progressive motor symptoms developed years before the cognitive symptoms developed could also help identify a unifying condition that might have spread throughout the nervous system. These considerations require keeping in mind gradually progressive neurological disorders that are associated with cognitive and pyramidal or extrapyramidal motor symptoms and have insidious onset in adulthood (
Table 1), while establishing the specific nature of the patient’s cognitive and motor changes by examination. Is the pattern of cognitive deficits suggestive of a neurodegenerative syndrome, or is it a relatively nonspecific pattern? Do the motor symptoms reflect true neuromuscular weakness, resulting from conditions involving the pyramidal pathway, neuromuscular junction, or muscles, or do the symptoms reflect elements of an extrapyramidal disorder that might affect fine motor dexterity, speed, or postural stability?
This patient’s cognitive examination was notable for a score of 24 of a maximum 30 points on the Montreal Cognitive Assessment (scores ≤25 indicate a level of impairment), with points lost for two errors in the serial seven subtraction task and a score 0 of 5 points on a delayed word recall task (although all five words were identified with category cues). A neuropsychological evaluation completed prior to referral documented average performance on tests of visual and spatial functioning, average to below average performance on tests of processing speed and executive functioning, low average performance on tests of basic attention and semantic fluency (with average phonemic fluency and confrontation naming), and exceptionally low word-list learning. Specifically, on a 12-item word-list test, the patient acquired five, six, and five words over three trials and recalled two of the 12 words after a delay (recognition results are not reported).
Pertinent findings from the patient’s general neurological examination included a resting tremor of the jaw, with no additional tremors, masked facies, or bradykinesia; normal muscle tone and no percussion myotonia; presence of notable atrophy of the forearm flexor compartments and quadriceps bilaterally and mild proximal arm atrophy; no fasciculations; and diffuse, relatively symmetrical weakness that was most pronounced in the finger flexors and hip flexors. There were no pertinent somatosensory deficits. Deep tendon reflexes were trace throughout, and plantar responses were flexor. The patient walked with a stooped posture, assisted by a cane, and he was unable to walk on his heels or toes. The Romberg sign was not present.
Questions: How does information from the neuromuscular and cognitive examination help to narrow the differential diagnosis? What diagnostic tests and studies are indicated?
The most salient result from cognitive testing was the patient’s impaired word-list learning, particularly on the 12-item task with a list length that exceeds working memory span. Poor retention—computed as the number of words obtained on delayed recall divided by the maximum number of words obtained on any of the acquisition trials—suggests temporolimbic amnesia, which is in line with the reported history of difficulties remembering recent events and information (
1). Taken together with borderline impairment in semantic fluency that was consistent with the reported history of difficulties retrieving words, this cognitive profile would most commonly be seen among those with Alzheimer disease (AD) (
2).
The presence of neuromuscular weakness, atrophy, hyporeflexia, normal muscle tone, and no pertinent somatosensory deficits suggests a disorder affecting lower motor neurons or the muscles themselves. The absence of bradykinesia, rigidity, and hyperkinetic signs, such as limb tremor, dystonia, or dyskinesias, makes diseases involving the basal ganglia less likely. Conditions in the differential diagnosis include motor neuron disease (MND) and myopathies. The lack of fasciculations or accompanying signs of upper motor neuron dysfunction argues against MND. The distribution of prominent atrophy of the quadriceps and medial forearm flexors and weakness in wrist and finger flexion and knee extension in an older patient raises the possibility of sporadic inclusion body myositis (IBM), an inflammatory myopathy (
3,
4).
Further workup is required to increase diagnostic confidence. Electromyography (EMG) and nerve conduction studies are indispensable for distinguishing between disorders of the motor neuron, nerve, neuromuscular junction, and muscle (
5). Findings suggestive of MND would include evidence of widespread acute denervation (fasciculation potentials, fibrillations, and positive sharp waves), chronic reinnervation (motor unit potentials [MUPs] exhibiting increased amplitude, increased duration, and polyphasia), and a reduced recruitment pattern (reflecting a loss of motor units). Findings suggestive of myopathy would include polyphasic MUPs with decreased amplitude and duration and an early recruitment pattern (reflecting an unchanged number of motor units that are activated earlier than expected because the number of normally functioning muscle fibers is reduced). In the case of inflammatory myopathies, including IBM, EMG can also demonstrate “irritable” myopathic changes characterized by increased insertional and spontaneous activity with fibrillation potentials and positive sharp waves, although these findings are not specific (
6).
A laboratory evaluation for muscle weakness should include a comprehensive metabolic panel and testing for calcium, magnesium, phosphate, creatine kinase, aldolase, lactate dehydrogenase, and thyroid-stimulating hormone levels (
7). Given the possibility of IBM, testing for anticytosolic 5′-nucleotidase 1A (anti-NT5C1A) antibodies is recommended (
8–
10).
Additional recommended evaluation workup for cognitive dysfunction would include MRI of the brain and an assessment of vitamin B12 levels (
11).
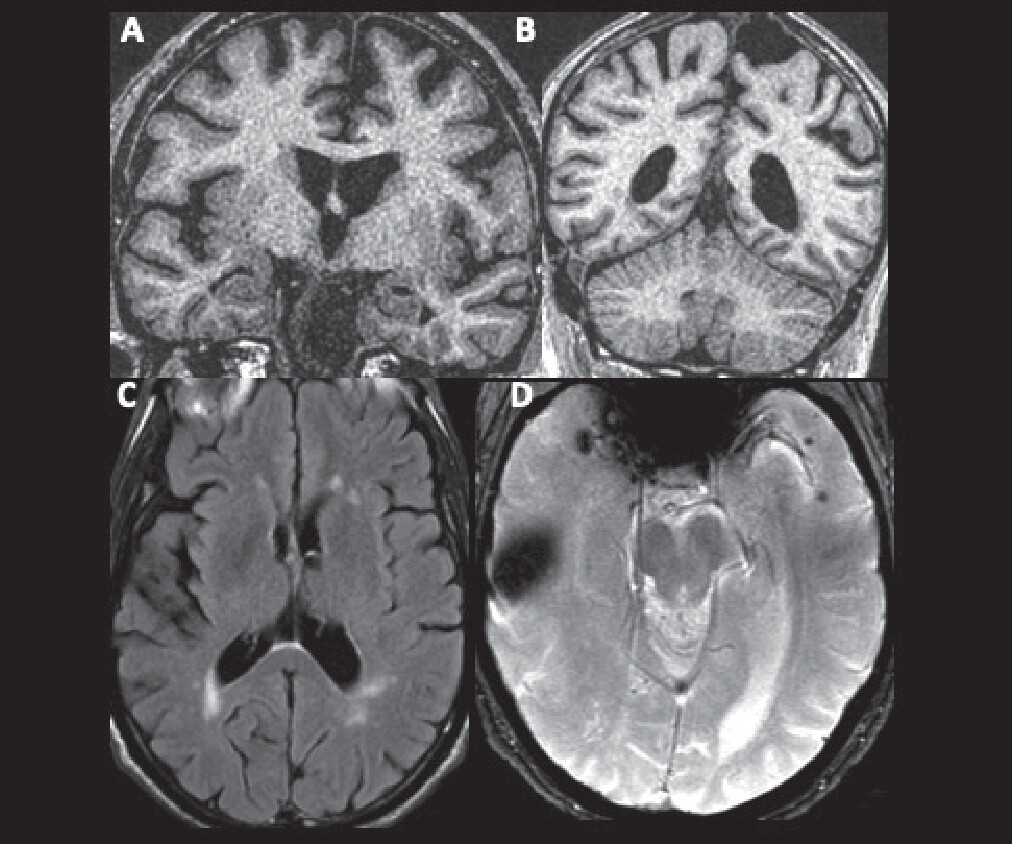
Results of the patient’s laboratory, diagnostic, and imaging tests are summarized in
Box 1 and
Figure 1.
Questions: Considering the results of these tests, what initial diagnostic formulation would be appropriate? Is a parsimonious, single underlying condition likely?
A diagnosis of amnesic mild cognitive impairment (MCI) possibly associated with mixed AD and vascular neuropathology is supported by the patient’s cognitive profile (involving early amnesia, borderline impaired lexical and semantic retrieval, and variable attention and executive functioning), the insidious onset and gradually progressive time course of symptoms, the preservation of the patient’s functioning in instrumental activities of daily living, and brain MRI that revealed evidence of small vessel ischemic disease and potential atrophy of the medial temporal, posterolateral temporal, dorsal frontal, and parietal lobes. The size of the focus of susceptibility (reflecting hemosiderin deposition) in the right anterior temporal lobe is consistent with the patient’s reported history of traumatic contusion. Multiple microhemorrhages in the left anterior temporal cortex could be consistent with traumatic axonal injury or cerebral amyloid angiopathy (CAA); either of these, in addition to the contusion, could contribute to cognitive impairment (
12,
13).
The EMG findings of short-duration, low-amplitude MUPs with early recruitment suggest a myopathy rather than MND; increased insertional activity, fibrillation potentials, and positive sharp waves further suggest “irritable myopathy.” The mildly but not substantially elevated creatine kinase level argues against dermatomyositis and polymyositis, as does the lack of proximal arm weakness. Factoring in the patient’s age, the specific distribution of weakness, and the time course of insidious onset and gradual progression of symptoms, these EMG and laboratory results support a diagnosis of sporadic IBM. Although associated with sporadic IBM, anti-NT5C1A antibodies have reported diagnostic sensitivity for IBM, ranging from approximately 40% to 75% across studies (
14,
15). However, their absence does not exclude the diagnosis, especially in the setting of an otherwise highly suggestive presentation.
A parsimonious, single cause of both the motor and cognitive symptoms appears less likely at this point. Mixed AD and cerebrovascular disease is not associated with focal muscle atrophy or myopathic EMG findings, even in later disease stages. Mutations in selected genes, including valosin-containing protein (VCP), cause hereditary multisystem proteinopathy (MSP) with heterogeneous phenotypes characterized by frontotemporal dementia (FTD), Paget disease of bone, myopathy with histopathological features similar to IBM, and, less frequently, amyotrophic lateral sclerosis (
16,
17). However, the distribution of weakness in MSP does not typically correspond to the canonical pattern in sporadic IBM, and no cognitive or behavioral features in this case suggest FTD. Myotonic dystrophy type 1 (DM1)—an autosomal dominant disorder caused by a trinucleotide repeat expansion in the dystrophia myotonica protein kinase gene—also warrants consideration in cases of adult-onset, gradually progressive neuromuscular weakness and cognitive dysfunction. Classic DM1 becomes symptomatic during the fourth decade of life at the latest, most commonly produces weakness in facial muscles, the sternocleidomastoid muscle, ankle dorsiflexors, and distal muscles of the forearms and hands, and is associated with cognitive changes that predominantly involve attention and executive dysfunction (
18–
20). These features and the lack of clinical or electrical myotonia make DM1 extremely unlikely in this case.
Question: What are therapeutic and prognostic considerations in this case?
AD and IBM are both gradually progressive diseases for which pharmacological treatments typically have modest benefits, at best. Consideration could be given to starting a cholinesterase inhibitor for cognitive impairment or deferring until the patient progresses to a stage of mild dementia when efficacy is more consistent (
21). While effective in dermatomyositis and polymyositis, immunotherapies have not been found to have meaningful benefit for individuals with IBM (
22). Nonpharmacological interventions for IBM are geared to optimize muscle strength and function in usual activities and reduce the risk of falling and aspiration, and these interventions include physical therapy, occupational therapy, speech therapy, exercise programs, and nutritional supports. Consultation with a pulmonologist or respiratory therapist can be useful to monitor the need for noninvasive ventilation, and consultation with a social worker can be useful to optimize caregiving support and resources (
23).
AD and IBM are both characterized by phenotypic heterogeneity with respect to rates of symptom progression, as well as the specific profile of symptoms (
3,
24,
25). For many patients, the most salient changes and greatest functional limitations during the mild to moderate stages of illness are related to progressive worsening of the more prominent symptoms at presentation. For this patient, progressive worsening would involve problems related to declining memory, dysphagia, and difficulties walking.
In the year following the initial presentation, our patient engaged with physical therapy, speech therapy, and otolaryngology services. He was hospitalized twice within a period of 3 months for aspiration pneumonia. He presented to the emergency department a third time when he tripped over a mat and hit the left side of his head on a desk, one of multiple falls in the interval since the evaluation. He continued to walk with a quad cane. His memory difficulties worsened such that he had difficulty placing recent significant life events in time and became more reliant on his wife to relay his history. He stopped driving, and his wife assumed responsibility for managing the family finances and overseeing his medications.
On neuropsychological reevaluation approximately 1 year after presentation, he was noted to be “bradyphrenic” and “tangential to a degree that he sometimes never answered the questions asked of him.” On testing, he exhibited “severe impairment in memory, language (semantic fluency), and executive functioning (working memory, set-shifting, processing speed)…(with) simple attention and visuoconstruction well below his baseline ability (but) areas of relative preservation.” He was started on donepezil, but this was discontinued shortly thereafter because of ongoing poor appetite and weight loss.
One and a half to 2 years after presentation, it was noted that he was very likely to sleep during the day when not actively engaged by others, despite sleeping 10–12 hours nightly using CPAP. He walked with a rollator walker outside of the home and independently within his apartment. He took 2–3 hours to finish a meal, adhering to a dysphagia diet supplemented with three to four cans of a high-calorie nutritional supplement daily. He was noted to be “not unhappy,” though without interest and motivation to engage in activities apart from watching television and listening to concerts at the assisted living residence where he and his wife had moved. He scored 11 out of 28 points on a modified version of the Mini-Mental State Examination (lower scores indicate greater impairment) that did not include drawing or writing tasks.
Notable changes following this visit included progressive daytime sleepiness, increased confusion in the evening, and transition to 24-hour care assistance. The patient died at age 77 from aspiration pneumonia, 12.5 years after he first experienced difficulties swallowing, 4.5 years following onset of memory difficulties, and approximately 2.5 years after presentation.
Discussion
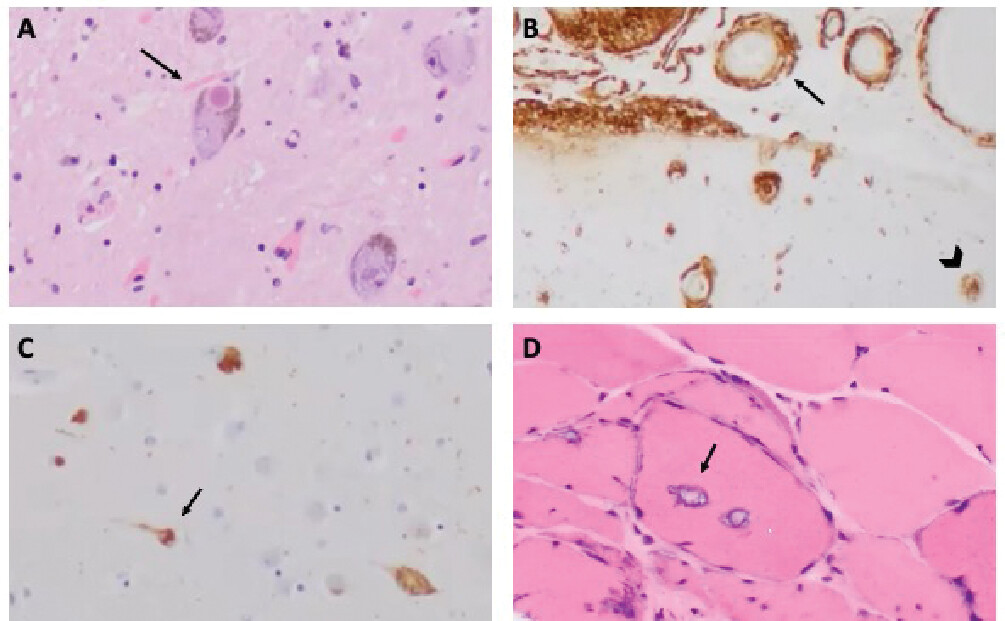
These neuropathological findings raise several interesting questions, including whether there were clinical features suggesting LBD prior to death, how various factors may have contributed to the accelerated clinical course of our patient’s cognitive, motor, and functional decline, and whether common pathophysiological mechanisms might give rise to the simultaneous development of advanced AD, LBD, CAA, and IBM.
In retrospect, certain clinical features, such as jaw tremor, stooped posture, bradyphrenia, tangential thought process, and excessive daytime somnolence despite adequate sleep overnight and adherence with CPAP, represent clues to the possibility of LBD. The absence of cardinal features of parkinsonism (bradykinesia, rest tremor, and rigidity), formed visual hallucinations, dream enactment (by history and preserved atonia during rapid eye movement sleep on polysomnogram completed shortly prior to presentation), and marked fluctuations in cognition disqualifies a diagnosis of either probable or possible dementia with Lewy bodies (DLB) by consensus clinical diagnostic criteria (
29). This dissociation between clinical symptoms and pathological findings illustrates the imperfect sensitivity of clinical diagnostic criteria for underlying neuropathology. Clinical and pathological studies of diffuse neocortical LBD pathology suggest a diagnostic sensitivity of approximately 75%–85% for the clinical diagnosis of probable DLB, which is reduced to 70% in cases with diffuse LBD and intermediate to high (Braak stages IV–VI) tau pathology (
30,
31).
This patient’s progression from MCI to moderate dementia over the course of 2 years was atypically rapid for AD (
24). His death from aspiration pneumonia 12–13 years after onset of neuromuscular weakness and 2–3 years following diagnosis of sporadic IBM was likewise atypical for IBM. IBM is associated with no effect on life expectancy among a majority of patients, although premature death due to aspiration pneumonia more than 15 years from the onset of symptoms and more than 10 years from diagnosis can occur among a subset of patients with more prominent early dysphagia (
32,
33). The presence of comorbid AD, LBD, and arteriolosclerosis, which is common in this patient’s age range (
34), likely accelerated his cognitive decline given evidence that LBD and cerebrovascular disease are independently associated with faster decline when comorbid with AD compared with AD occurring in isolation (
35,
36). His history of traumatic brain injury (TBI) with cerebral contusion and subarachnoid hemorrhage also potentially contributed to rapid progression, with research suggesting faster progression of functional impairment among patients with AD and a history of TBI within 10 years of symptom onset, compared with patients with no history of TBI (
37). Additionally, the combination of apathy related to neurodegenerative disease and dysphagia related to IBM could have contributed to the patient’s decline in cognitive and functional status, which aligns with evidence that nutritional factors influence rates of symptomatic progression and quality of life not only among individuals with dementia but also among those with myopathy and systemic inflammatory disorders (
38–
40).
An epidemiological association between AD, LBD, and IBM has not been established beyond rare case reports of patients with AD and IBM (
41,
42). Citing similar histopathological features of AD, LBD, and IBM, including aggregates containing beta-amyloid, phosphorylated tau (p-tau), or alpha-synuclein in IBM muscle fibers, some investigators have proposed similar upstream disease mechanisms centered around the cytotoxicity of misfolded protein aggregates (
43). However, in contrast to AD and LBD, IBM muscle fibers have been associated with the accumulation of dozens of additional aggregated molecules, and beta-amyloid, p-tau, and alpha-synuclein inclusions are present less consistently than p62, light chain 3, and transactive response DNA-binding protein of 43 kDa (TDP-43)—inclusions that reflect disruptions in the RNA metabolism response to stress and alterations in autophagy, the major intracellular degradation system by which old, damaged, or abnormal cytoplasmic materials are delivered to and degraded in the lysosome (
3,
26,
27,
44). More importantly, immunological mechanisms appear to play a more consistent upstream role in IBM, evidenced histologically by the invasion of myofibers by cytotoxic T cells (a “signature” of highly differentiated CD8 T-cell effector memory and terminally differentiated effector cells), identification of anti-NT5C1A antibodies, strong genetic linkage of IBM to human leukocyte antigen gene variants associated with autoimmune disease, and microarray detection of abundant immune chemokine and cytokine transcripts in muscles affected by IBM (
3,
45).
Mutations in genes including VCP and sequestosome 1 (the gene that encodes p62) cause MSP with histopathological features of myopathy resembling sporadic IBM but usually without inflammatory changes. This situation raises a question of whether a minority of cases of sporadic IBM without anti-NT5C1A antibodies and lymphocytic infiltrates could be driven predominantly by RNA stress granule dysfunction and impaired autophagy (
16). These mechanisms have been implicated not only in frontotemporal lobar degeneration but also in AD and LBD (
46–
50). If neurodegeneration hinges on both cellular stress and inadequate cellular response mechanisms, then sporadic neurodegenerative diseases could arise in the context of grossly excessive stress, dysfunctional stress-response mechanisms, or some combination of the two. In AD, numerous types of cellular stress have been implicated, including beta-amyloid toxicity in the context of overproduction or impaired clearance (
51), loss of presenilin function (
52), cerebrovascular disease (
53), impaired insulin signaling and other forms of metabolic stress (
54), neuronal hyperexcitability (
55), mitochondrial dysfunction (
56), immunological dysregulation (
57), infection (
58), environmental toxins (
59), and head trauma (
60). AD has high comorbidity not only with LBD pathology but also with TDP-43 (
61). It is plausible that in a case like the one discussed in this article, polygenic or unidentified genetic factors could confer vulnerability to multiple sources of age-related cellular stress to produce a “sporadic multisystem proteinopathy.” It is also plausible that our patient developed these multiple and substantial pathologies of the brain and muscle independently, for unrelated reasons.