Case Presentation
A very-high-functioning 72-year-old right-handed man raised concerns with a general neurologist about subtle memory difficulties and was told he likely had Alzheimer’s disease (AD). The patient openly shared this diagnosis with family members and friends, none of whom had noticed cognitive changes. Some thought the patient was exaggerating his problems, perhaps to garner sympathy and attention. Approximately two-and-a-half years later, his cognitive difficulties led him to retire as a practicing psychologist. “I could not remember what my patients had told me from week to week; even worse, I could not remember their names from one week to the next,” he explained during his first visit to a cognitive neurologist the following year at age 75.
The patient also complained of having difficulty following the content and flow of extended conversations and keeping track of routine tasks around the house. “I leave water running, leave my credit card after I pay a check …even leave my wallet if I happen to put it down. I left a car door open one time when I backed out of the garage. The door got smashed against the side of the garage exit.” He began writing himself notes that he taped on walls and doors. Although no longer able to work, he continued to handle the family finances and work on academic manuscripts (that were left unfinished). He stopped managing his stock market investments and became reliant on his wife to arrange social activities. His deteriorating cognitive status and increased dependence were associated with a worsening of his long-standing dysthymia.
His medical history included atrial fibrillation, coronary artery disease, hypertension, hyperlipidemia, major depression, Crohn’s disease (in remission), restless leg syndrome, and obstructive sleep apnea, which was treated with continuous positive airway pressure. His medications included warfarin, sertraline, famotidine, and vitamin E. He had no history of alcohol or recreational drug use. He quit smoking when he was in his 60s.
The patient earned a Ph.D. degree in clinical psychology and had served as a leader in professional organizations. His first marriage, which produced four children, ended in divorce. He had been married to his second wife (also a psychologist) for 40 years. There was no family history of neurodegenerative disease.
Question: What additional information would cognitive, neurological, and laboratory-based results provide?
Brief, initial office-based neurocognitive testing with a cognitive neurologist approximately 3 years after symptom onset revealed mild deficits in the memory and language domains. The patient’s basic attention and working memory were intact, with a digit span of eight forward and six in reverse. His verbal episodic memory, measured by using a seven-item word span task, was mildly impaired. He encoded 6, 7, and 7 words over three learning trials and spontaneously recalled all seven words after 30 seconds but recalled only two words after 3 minutes; on multiple-choice testing, he recognized an additional four words, without false positives. He had mild naming deficits for high-frequency items, such as “computer monitor.” He generated 22 “F words” (letter fluency) and 21 animals (category fluency), each in 1 minute. There was no evidence of ideomotor apraxia, agnosia, cortical sensory loss, pseudobulbar affect, or behavioral disinhibition.
A sensory-motor neurological examination revealed decreased reflexes throughout, a mildly wide-based gait, and difficulty with tandem gait. There were no signs of oculomotor dysfunction, bradykinesia, rigidity, postural instability, or focal sensory or motor deficits. In short, his examination did not suggest evidence of parkinsonism or focal deficits.
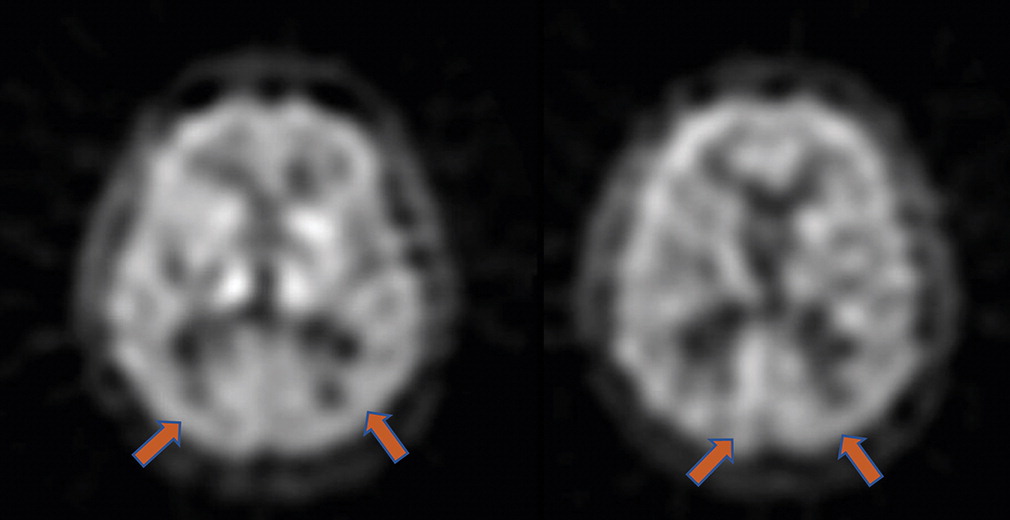
A laboratory workup for cognitive impairment, including vitamin B12 level and thyroid function tests, was unremarkable. The patient’s initial brain MRI, obtained during the year of symptom onset (at age 72), reportedly revealed mild diffuse atrophy without a pattern of regional cortical volume loss associated with a specific neurodegenerative disease (images are not available). No evidence of significant small-vessel disease or large-vessel strokes was reported. The patient also underwent single-photon emission computed tomography (SPECT) that showed mild bilateral hypoperfusion in temporal-parietal regions (
Figure 1).
Question: Given the workup to date, what would be an appropriate diagnostic formulation?
In summary, our patient was a 75-year-old man with very high cognitive reserve, whose symptoms included difficulties in learning and retrieving new information (e.g., recalling what the patients in his clinical psychology practice had reported in previous weeks) and carrying out goal-directed tasks without becoming distracted. On testing, he exhibited memory deficits, especially at the level of encoding and retrieval. He had difficulty rapidly retrieving the names of common objects, and in contrast to cognitively normal individuals (
1), his category fluency was not greater than his letter fluency (indicative of impairment in lexical-semantic information access). His brain MRI scan revealed mild global atrophy, and his SPECT scan demonstrated bilateral temporal-parietal hypoperfusion.
At a syndromic level, the patient exhibited multidomain cognitive impairment (memory, naming, semantic access, and executive control). Cognitive impairment led him to retire from work and become more dependent on his wife, which is consistent with dementia of mild severity. The slowly progressive cognitive impairment strongly suggested a neurodegenerative process, and his pattern of cognitive deficits and imaging findings were consistent with underlying AD as a likely cause. Although cerebrovascular disease (with risk factors of hypertension, hyperlipidemia, coronary artery disease, and atrial fibrillation), obstructive sleep apnea, and increased dysthymia may have contributed to his cognitive difficulties, they were unlikely to be the main source of his clinical syndrome.
Question: How does the patient’s view of his illness affect his choices and behavior?
Trials of donepezil and then galantamine were not tolerated due to complaints of dizziness; thus, memantine was started. During the year he turned 77, the patient abruptly announced that he did not want to have AD and would no longer accept the diagnosis, and therefore, he decided to discontinue his medications. He sought other medical opinions from other experts in the field to “prove that the diagnosis was wrong.” When most of these experts concluded that he had AD, he “gave in” and restarted taking memantine and sertraline. He struggled with worsening mood and apathy, prompting a neuropsychiatric assessment that led initially to an increase in his dose of sertraline and the subsequent addition of methylphenidate, which seemed effective.
Over time, the patient not only acknowledged having AD but also fully embraced the diagnosis. He became an articulate “spokesperson” for the regional Alzheimer’s Association and gave many prepared speeches at organized conferences that provided “the patient’s perspective.” According to his wife, this activity developed as a result of their participation in many support groups offered by the Alzheimer’s Association. She would attend a caregiver support group while he went to a patient-centered group. These patient groups would disband as the condition of its members deteriorated and they could no longer actively participate. However, her husband did not seem to get much worse, and he would join another, newly formed patient group. This scenario continued to repeat itself. After some time, staff at the Alzheimer’s Association got to know the patient and asked whether he would like to speak about his experience as a person with AD. Of note, the patient was featured on the front cover of the annual report of the regional Alzheimer’s Association.
Over the next 5 years, the patient exhibited a slowly progressive decline in cognitive abilities. The salient changes in his performance on neuropsychological testing (
2) between ages 76 and 82 are summarized in
Table 1. He exhibited a substantial decline in his performance on tests of memory (both delayed recall and recognition tasks), retrograde memory for U.S. presidents, verbal memory for contextual information, visual memory, naming, and category fluency. However, his performance on tests of executive functioning, attention, and visuospatial skills were essentially unchanged. His sensory-motor neurological examination was stable and did not reveal focal weakness, bradykinesia, rigidity, tremor, postural instability, or oculomotor abnormalities. Functionally, although the patient continued to manage his basic activities of daily living (ADLs), he exhibited a progressive deterioration in instrumental ADLs. For example, his wife took over the family finances and discouraged him from driving. He stopped initiating activities and became increasingly dependent on his wife, who supervised his daily affairs.
When the patient was in his mid-80s, approximately 10 years after symptom onset, dementia severity remained in the mild range, and he scored 24 out of 30 on the Mini-Mental State Examination (MMSE). With his wife’s support, he volunteered to participate in an AD clinical drug trial investigating solanezumab, an experimental antiamyloid antibody medication. To confirm trial eligibility, during screening, he underwent a research-based [
18F]florbetapir amyloid positron emission tomography (PET) study. The scan was interpreted as negative for amyloid (i.e., the standardized uptake value ratio was below the trial’s cutoff for amyloid). Because abnormal amyloid deposition is required for a diagnosis of AD and because the radiological-pathological concordance between florbetapir PET results and fibrillar amyloid deposition is high (
3,
4), the PET study suggested that the patient did not have AD.
Question: How do the patient and his care team make sense of an unexpected neuroimaging result?
Having come to terms with and then fully embracing the diagnosis of AD, the patient and his wife were initially perplexed by this turn of events. He was no longer experiencing the illness his doctors had diagnosed more than 10 years earlier. His wife was very disappointed that he was not eligible to receive a “promising therapy” for his dementia. On the other hand, the patient quickly became delighted that he “no longer had” AD.
Members of his clinical neurology team were also surprised by the negative amyloid PET results. They considered investigating cerebrospinal fluid biomarkers of AD (amyloid-beta 42, total tau, and phospho-tau) because some studies have suggested discordance between biomarker indicators and amyloid PET results in up to 20% of cases (
5,
6). However, after discussions with the patient’s wife, the team decided not to pursue a lumbar puncture, because it would have necessitated discontinuation of anticoagulation therapy for atrial fibrillation, putting him at risk for a stroke.
Question: If the patient does not have biomarkers consistent with AD, what is the underlying illness?
Before the availability of large autopsy series and in vivo biomarkers of AD neuropathology, a prevailing view was that amnestic dementia could be considered synonymous with AD, in accordance with consensus clinical diagnostic criteria for “probable AD” (
7). Subsequent large clinical and neuropathological studies suggested that a clinical diagnosis of “probable AD” made by experts had a positive predictive value of approximately 83%–90% for intermediate to high levels of AD neuropathology, defined by consensus neuropathological criteria (
8,
9). Similarly, early studies employing amyloid PET suggested that clinically diagnosed “probable AD” predicted elevated brain amyloid with a positive predictive value of approximately 82%–86%. However, approximately 35%–40% of noncarriers of the apolipoprotein E ε4 (
APOE ε4) allele who were diagnosed with “probable AD” did not have elevated amyloid deposition (
10,
11). These findings supported conceptualizing the in vivo diagnosis of AD on the basis of the biomarkers amyloid (A), tau (T), and neurodegeneration (N). In this framework, amnesic mild cognitive impairment or dementia and an A–N+ biomarker profile was indicative of “suspected non-Alzheimer’s pathophysiology” (SNAP) (
12,
13). This profile applied to our patient, who had a florbetapir PET study suggesting low amyloid deposition, as well as neurodegeneration suggested by atrophy on MRI and hypoperfusion on SPECT imaging.
A wide range of pathologies have been associated with SNAP, and these pathologies are often associated with inclusions containing misfolded proteins other than amyloid (e.g., tau, alpha-synuclein, and transactive response DNA-binding protein 43 [TDP-43]), with or without low levels of AD pathology. Amnestic dementia involving tau pathology without amyloidosis could include primary age-related tauopathy, argyrophilic grain disease, and less frequently, progressive supranuclear palsy or corticobasal degeneration (
9,
12,
14,
15). Pathologies of hippocampal sclerosis, cerebrovascular disease, or Lewy body disease, an alpha-synucleinopathy, can present initially with progressive memory impairment (
16,
17). Cerebrovascular disease is often accompanied by focal deficits on the sensory-motor neurological examination and more dominant executive dysfunction, whereas Lewy body disease is often accompanied by parkinsonism, fluctuations in arousal level, hallucinations, or rapid eye movement sleep behavior disorder (
16,
17).
Limbic-predominant age-related TDP-43 encephalopathy neuropathological change (LATE-NC), with or without hippocampal sclerosis, is another common neurodegenerative proteinopathy that predominantly affects individuals 80 years or older, can present with SNAP, and mimics the most common clinical presentations associated with AD. However, when our patient was diagnosed with SNAP, the type of dementia referred to as LATE had not been identified yet; LATE emerged as a classification in 2019 (
18).
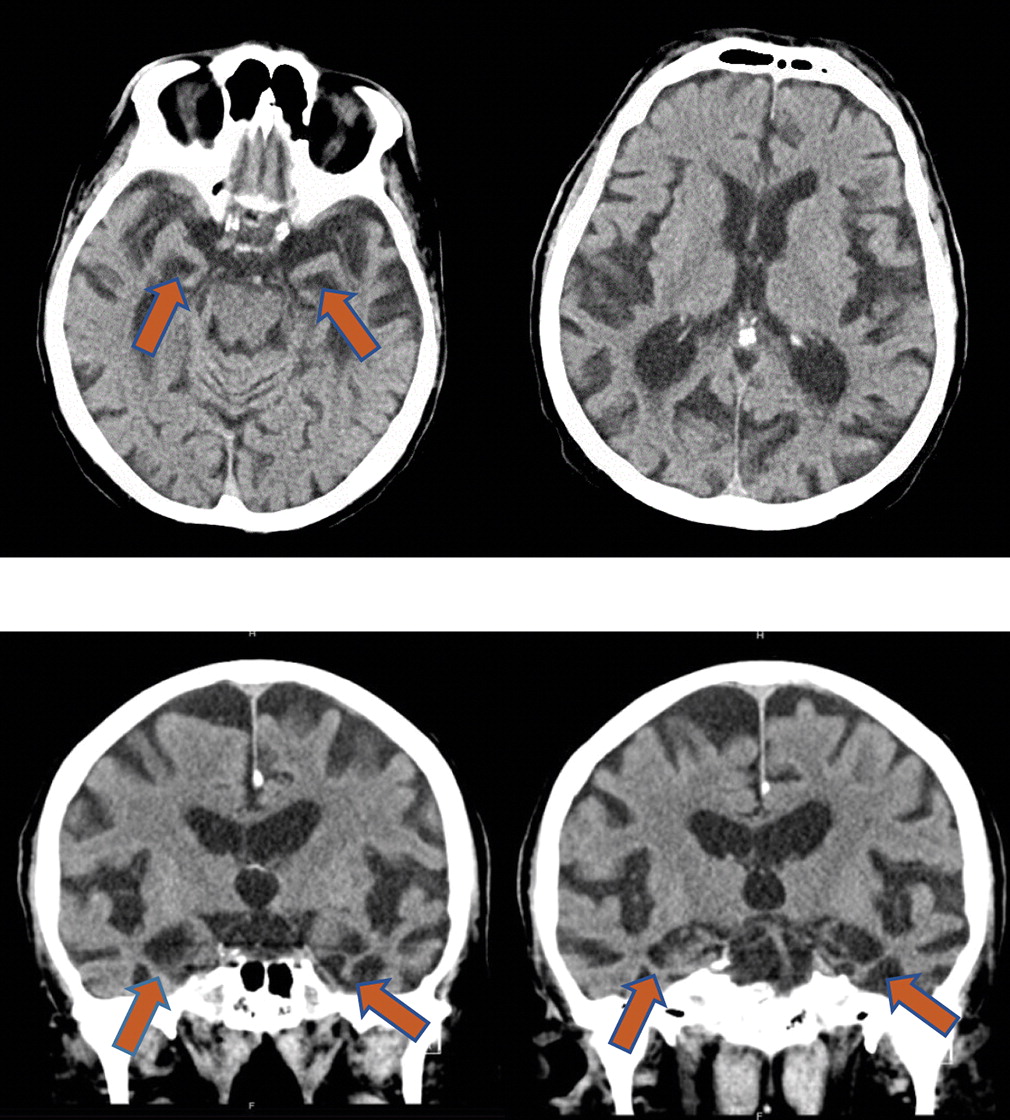
Approximately 17 years after our patient’s initial presentation to a neurologist and approximately 3 years after an amyloid PET study, he started to develop visual hallucinations, agitation, and gross disorientation to time and place. During a hospital admission the following year for neuropsychiatric symptoms, a head computed tomography scan revealed prominent hippocampal atrophy bilaterally (
Figure 2). He died a month after hospital discharge due to complications of aspiration pneumonia, and permission for an autopsy was granted.
Discussion
Our patient’s slowly progressive amnestic, multidomain cognitive impairment in his mid-70s, along with imaging scans that revealed evidence of cortical atrophy and temporal-parietal hypoperfusion, led to a clinical diagnosis of dementia of the Alzheimer type. His documented deterioration in memory, naming, and semantic processing between ages 76 and 82 (
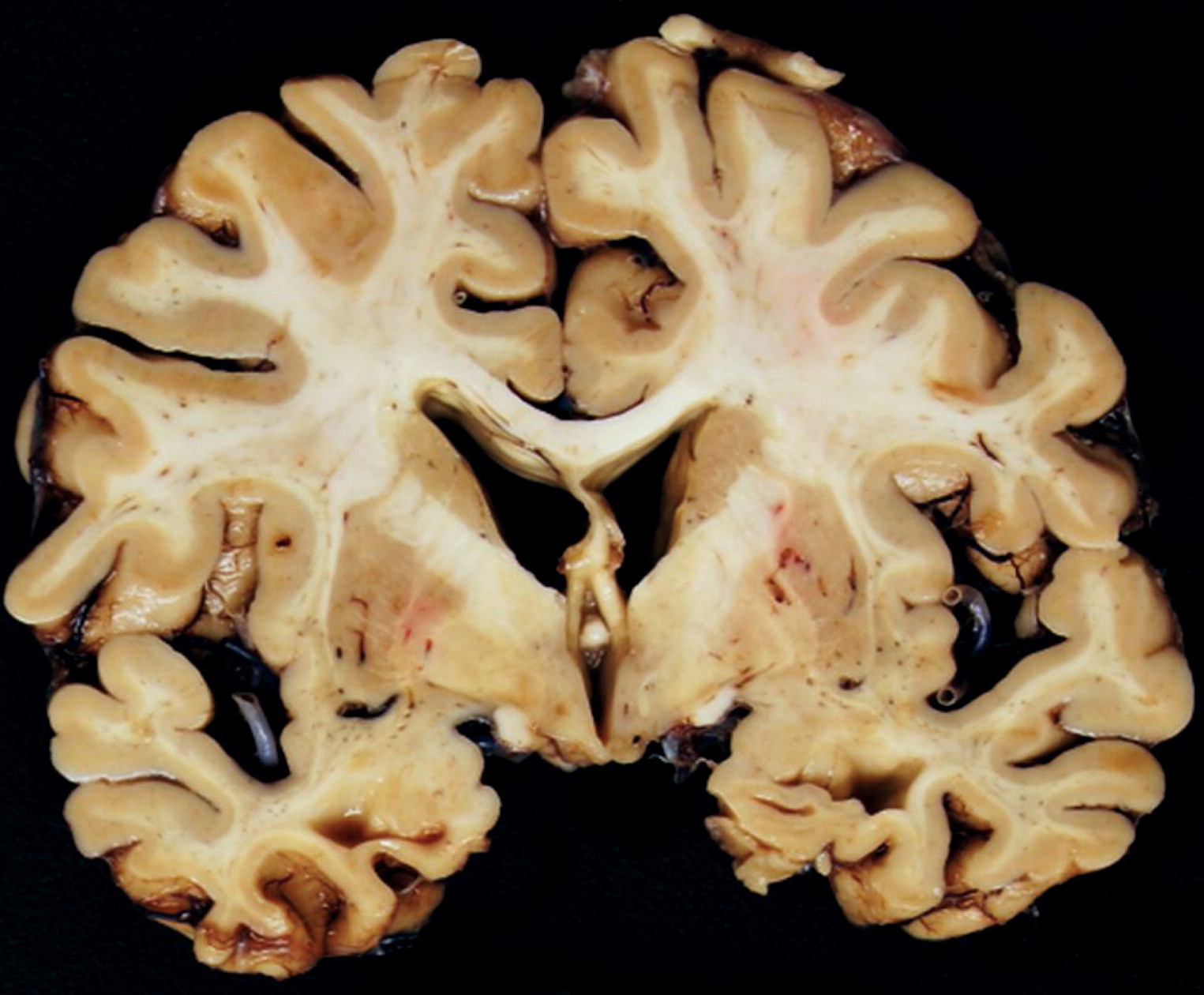
Table 1) strengthened this formulation. However, after more than 10 years of carrying the diagnosis of AD, an amyloid PET study was interpreted as being negative, leading to the conclusion that in fact, this patient did not have AD. Approximately 3 years later, an autopsy was performed, which included a pathological diagnosis of AD. The challenge is to understand how this patient had, then did not have, and then did have AD and to determine whether AD pathophysiology was the primary cause of his clinical syndrome.
How should we interpret the discordance between the patient’s amyloid PET findings and the autopsy results? One possibility is that the AD pathological process started after age 85, when the amyloid PET study was conducted. This explanation seems unlikely because amyloid accumulation usually develops over the course of decades. Another possibility is that the amyloid PET study yielded a false-negative result, which has been shown to occur in a small percentage of cases in which autopsies demonstrated fibrillar amyloid plaques (
19,
20). We believe that the most plausible explanation is that when this patient was 85 years old, fibrillar amyloid had started to accumulate but was below the threshold value for being interpreted as positive in the amyloid PET study (
19). Notably, the quantitative thresholds using standardized uptake values from PET studies for determining elevated levels (i.e., a positive result) or for indicating levels that are not elevated (i.e., a negative result) in AD clinical trials are variable.
If we assume that our hypothesis is correct and that amyloid had been slowly accumulating, there are several reasons that AD was unlikely to be the primary neuropathological process driving our patient’s clinical course. The pace of his decline would have been quite slow for typical AD and even for limbic-predominant AD, a neuropathologically defined subtype of AD characterized by a relatively higher burden of neurofibrillary tangles in the hippocampus, lower burden of neurofibrillary tangles in neocortical areas, later mean age at onset, and slower mean rate of progression compared with typical AD (
21). For example, more than 10 years after the start of the patient’s clinical illness, his MMSE score was 24 out of 30 points, and after more than 15 years, his autopsy revealed only an intermediate degree of AD neuropathology, as indexed by Braak stages III–IV/VI (
22). Studies have suggested that for any given level of cognitive impairment, individuals with high cognitive reserve (such as our patient) demonstrate a higher burden of amyloid than those with lower cognitive reserve (reflecting the ability of a patient with high cognitive reserve to compensate for disease load) (
23). Thus, if AD pathology had been the primary cause of the patient’s 10-plus years of progressive cognitive and functional decline, we would have expected an amyloid load sufficient to have led to a positive amyloid PET outcome. The relatively lower AD neuropathological change (ADNC) burden found at autopsy could not fully explain this patient’s clinical presentation.
Instead, we believe that the primary neuropathological process in this patient’s case was LATE-NC. Although TDP-43 proteinopathy was first described in frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) in 2006 (
24), neuropathology laboratories across the world soon recognized that TDP-43 proteinopathy is also very common among older adults without FTLD or ALS and is associated with amnestic cognitive decline. This new clinicopathological entity was named LATE in 2019, and the associated TDP-43 proteinopathy was named LATE-NC. LATE-NC is by far the most common type of TDP-43–related neurodegeneration, more than 100 times more common than FTLD (
18).
In both imaging studies during life and neuropathological evaluation at autopsy, LATE generally reveals disproportionately greater hippocampal volume loss compared with other regions (
18,
25,
26). In contrast, imaging studies among patients with AD tend to demonstrate a much wider variation in the brain regions affected, which include the hippocampi, parietal lobes, and temporal neocortex (
27). Although it is common for patients with LATE to exhibit gliosis in the hippocampi, consistent with hippocampal sclerosis (
18), LATE can present with or without hippocampal sclerosis.
LATE and, more generally, TDP-43 proteinopathies should not be considered rare conditions, because TDP-43 inclusions are very common in older populations (
18,
28). LATE-NC affects more than 25% of individuals in their ninth decade and beyond and is the third leading pathology associated with dementia, following ADNC and cerebrovascular disease. There is evidence that LATE-NC accounts for approximately 15%–20% of clinically diagnosed AD in advanced age (
18). One challenging aspect of LATE-NC is that it is often accompanied by AD pathology. Between 20% and 50% of pathologically proven cases of AD and 75% of cases involving a severe degree of pathology (Braak stages V and VI) also exhibit TDP-43 abnormalities (
29).
Furthermore, when present with ADNC, LATE-NC is associated with faster cognitive decline, greater hippocampal atrophy, and higher risk of dementia and is a critical determinant of clinical prognosis among those with AD (
30). Nevertheless, approximately 15% of LATE-NC cases are found among older adults with no or low ADNC burden. Because LATE-NC predominantly affects temporolimbic structures, patients with LATE-NC present with amnestic cognitive decline that resembles the AD clinical syndrome, even in the absence of biomarker evidence of AD (i.e., SNAP) (
18). In LATE-NC cases not accompanied by ADNC, cognitive decline is slower, and impairments beyond episodic memory are less apparent (
31,
32).
Genetic studies have shown that common risk variants (
28,
33–
35) associated with AD (
APOE ε4) and FTLD-TDP (
TMEM106B rs1990622
A and
GRN rs5848
T) are also seen in LATE-NC, suggesting overlapping mechanisms across these entities. Furthermore, a brain gene-expression study (
36) showed that dysregulation in lysosomal function may underlie both ADNC and LATE-NC. Nonetheless, the pathophysiology of LATE-NC remains largely unknown.
With the advent of disease-modifying drugs, clinical practice is likely to rely on a growing number of AD biomarkers, which currently include amyloid PET results and amyloid and tau protein levels in cerebrospinal fluid and, relatively soon, in blood (
37). Patients, especially those in their 80s and 90s, with clinical presentations suggestive of AD and negative tests for AD biomarkers may have underlying TDP-43 pathology and LATE. Unfortunately, there is no biomarker available at this time to determine the presence of TDP-43 during life; the diagnosis can only be confirmed by pathological investigation. LATE should be seriously considered among older patients, especially those in their 80s and 90s with memory impairment who exhibit slow progressive decline. Imaging findings (as provided in
Figure 2) that reveal disproportionate bilateral hippocampal atrophy would support this diagnosis. Negative AD biomarkers would increase suspicion for LATE, although given the prevalence of mixed pathologies, positive AD biomarkers would not rule it out. For example, some individuals classified as having SNAP because of their AD biomarker status likely have a TDP-43 proteinopathy such as LATE. Previous reports of LATE pathology, typically presenting with salient neuropsychiatric symptoms or with motor or focal sensorimotor symptoms, have not been substantiated (
38).
As biomarkers for underlying neurodegenerative diseases become more readily available, reliable, and reasonably priced (e.g., through blood work), their routine use in clinical settings is likely to increase substantially. Patient diagnoses based only on clinical assessment and standard evaluations, which occurred for many years with the patient reported herein, will need to be framed “provisionally,” because the diagnoses may require updating after biomarkers are obtained. This case highlights how important “labeling” a disease can be to patients and their families as they try to make sense of an illness that profoundly affects their lives. Moving forward, clinicians will be tasked with explaining several emerging, nuanced concepts to their patients. For example, although traditionally used categorical approaches to diagnosing a disease (i.e., the disease is either present or absent) seem straightforward and may reduce the discomfort of diagnostic uncertainty, such approaches can obscure the dimensional nature of the pathophysiological process (e.g., amyloid accumulation is on a continuum, with results close to but below cutoff values indicative of accumulating disease pathology). Although biomarkers may improve diagnostic accuracy, compared with the gold standard of neuropathology, that accuracy is far from perfect. On the basis of postmortem studies, biomarkers will likely be positive for a mixture of underlying neuropathologies that can be more challenging for patients to understand their condition and to find peer and community support associated with a specifically named illness.
The landscape of views about dementia seemed much simpler in the past. For much of the 20th century, dementia in old age was believed to be due to hardening of the arteries. By the 1980s, the pathology identified by Dr. Alois Alzheimer in 1906, which had been thought to reflect a rare presenile dementia, turned out to be the same pathology as that observed among older patients with dementia. The diagnostic pendulum swung strongly toward AD, which accounted for a very large portion of dementia cases. The pendulum seems to be shifting again, now toward an appreciation that many pathophysiologic processes can contribute to a patient’s cognitive, behavioral, and functional decline, with concomitant proteinopathies and mixed vascular and degenerative disease representing the rule rather than the exception (
39). It is hoped that in the future, medical practice will be positioned to identify biomarkers for all pathological protein inclusions, understand fully the various mechanisms giving rise to them, and employ a comprehensive array of disease-modifying therapies to target the specific mechanisms applicable for any given individual (
40).