Canst thou not minister to a mind diseased? Pluck from the memory a rooted sorrow, Raze out the written troubles of the brain, And with some sweet oblivious antidote Cleanse the stuffed bosom of that perilous, Stuff which weighs upon the heart?
—William Shakespeare, Macbeth
Recent findings have substantially increased our understanding of the pathophysiology of depression. There has been a correspondingly significant increase in our understanding of the efficacy and tolerability of currently available treatments. The latter database has convincingly demonstrated a large unmet need for the more than one half of depressed patients who fail to achieve remission after an adequate trial of antidepressant monotherapy. Despite our increased understanding of both its pathophysiology and treatment, depression remains highly prevalent, accounting for more disability than any other disorder worldwide. In fact, rates of major depression in the United States rose markedly in the decade from 1991–1992 to 2001–2002, from 3.33% to 7.06% (
1). It is the most significant risk factor for suicide, a leading cause of death worldwide, especially in adolescents, young adults, and elderly individuals. Indeed, suicide is the third leading cause of death in children and adolescents, and childhood depression and bipolar disorder are not uncommon, yet quite understudied. Depression is also an important risk factor both for the development of cardiovascular disease including myocardial infarction and congestive heart failure (
2) and for the poor response to treatment in these patients.
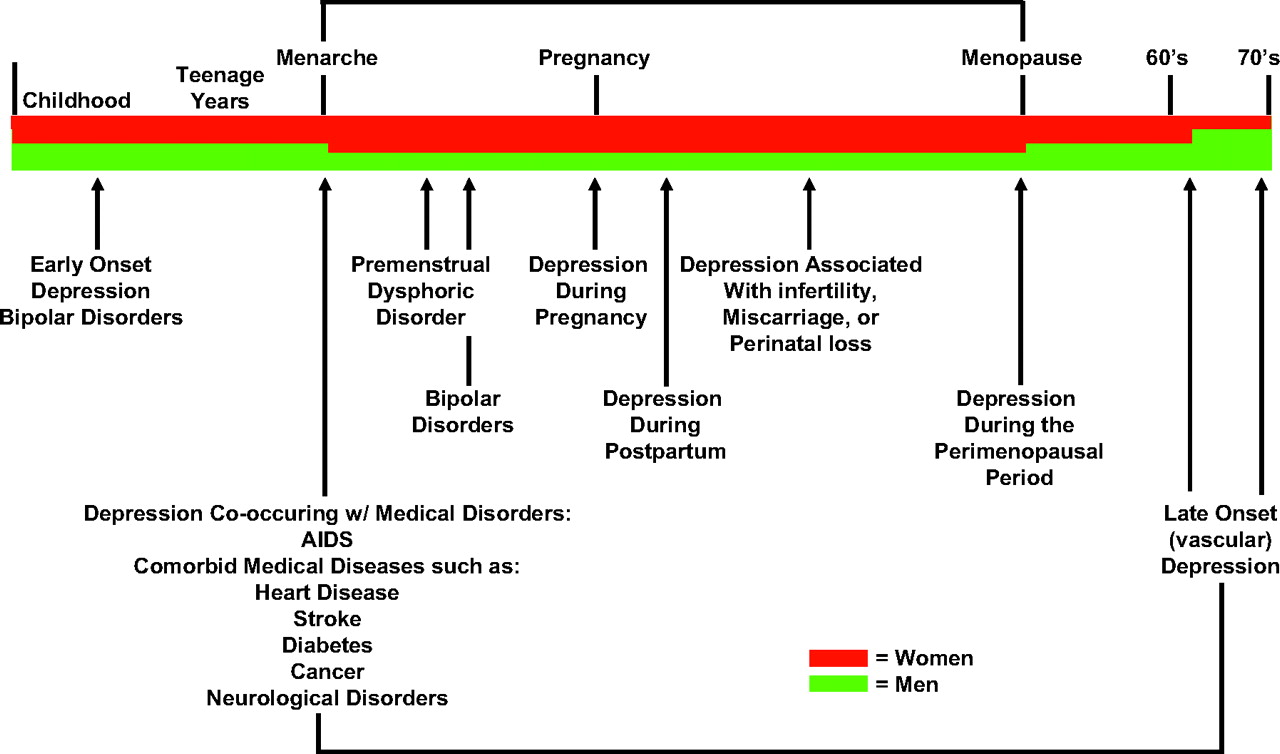
Figure 1 summarizes the major mood disorders across the life cycle and the diagnostic criteria for major depression.
There are still no validated, diagnostically useful biological tests for depression and none that reliably predict a response to one or another of the well-established and effective treatments for depression, except perhaps plasma drug concentrations of the older and side effect-prone tricyclic antidepressants. Similarly, although some promising candidates are emerging, there are no biomarkers, such as expression of a gene or tumor marker, that reliably change as a function of treatment response to antidepressants or psychotherapy (e.g., prostate-specific antigen is used to monitor efficacy of prostate cancer treatment).
Of considerable concern is the relatively recent realization stemming from several large-scale treatment studies, both of efficacy and effectiveness, that currently available antidepressants and psychotherapy, in particular cognitive behavior therapy (CBT), although clearly more effective than placebo, are as monotherapies associated with response (a 50% or more improvement in depressive symptoms) and remission (return to premorbid state) rates that are clearly unacceptably low. Combination or augmentation therapies comprised, respectively, of more than one antidepressant medication or an antidepressant and a second nonantidepressant drug to enhance the effects of the antidepressant, and combination pharmacotherapy/psychotherapy, although understudied, appear to be associated with better therapeutic responses than monotherapy. However, the increased side effects often, but not always, associated with coprescribing two medications, and the increased cost of treatment with combination psychotherapy and pharmacotherapy or two medications are major obstacles that prevent their wholesale clinical adoption. Second, with few exceptions, there is insufficient evidence of increased efficacy currently available to support a change in clinical practice. The results of the large-scale National Institute of Mental Health (NIMH)-sponsored clinical treatment trial, STAR*D, with its disappointing remission rates of 28%–33%, depending on the outcome measure, after up to 14 weeks of treatment with an selective serotonin reuptake inhibitor (SSRI), citalopram, highlight the paramount importance for further studies that seek to better understand the pathogenesis and pathophysiology of depression (
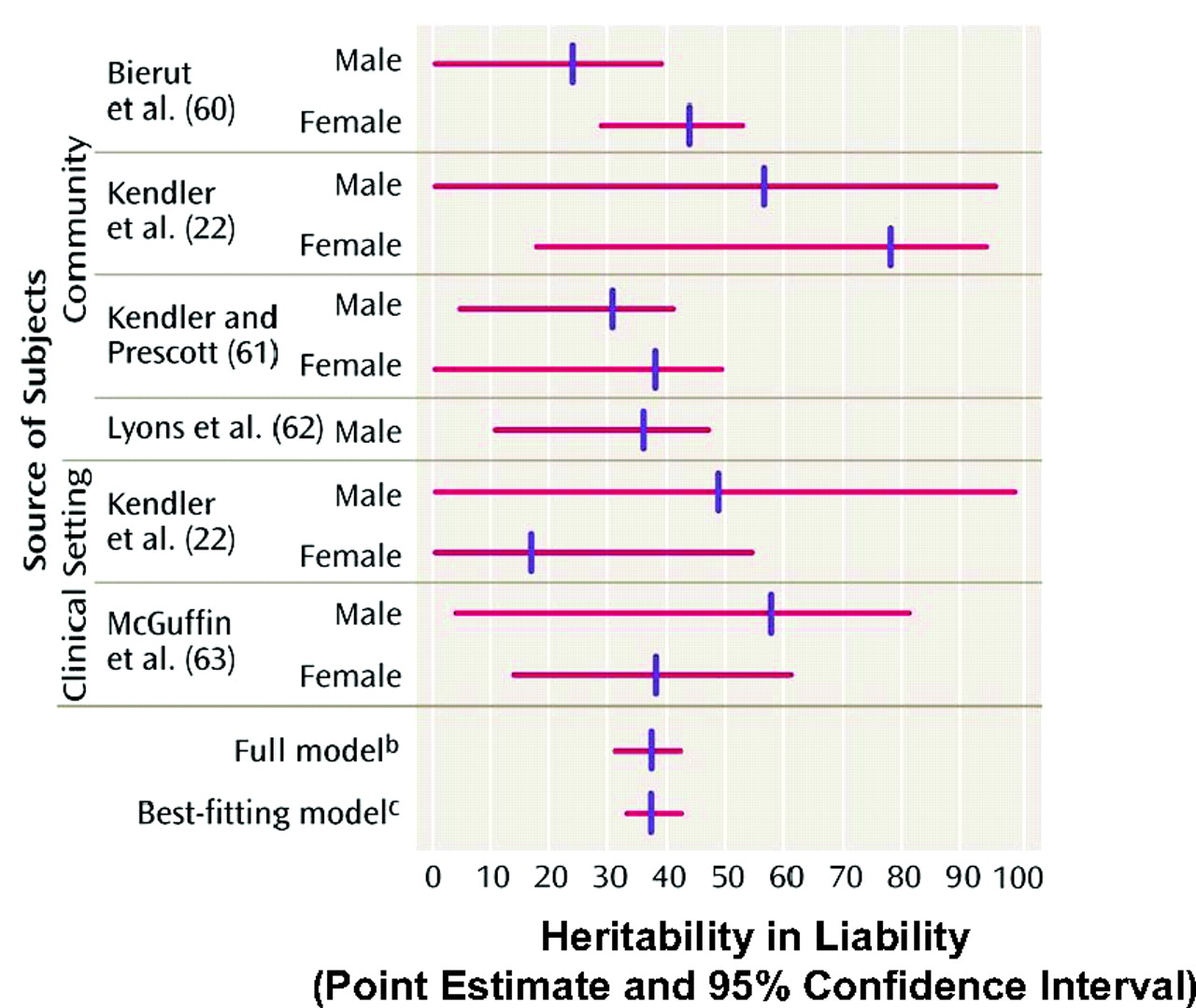
3). A comprehensive understanding of the genetic and environmental contributions to depression and its associated neurobiology is required before scientifically based, rational new treatment strategies are likely to be developed. Approximately one third of the risk for the development of depression is inherited (
Figure 2) and two thirds is environmental (
4). Many new findings and research directions that require immediate follow-up because one or more presage rapid changes in clinical practice have recently emerged—in diagnostics, the choice of currently available treatments, and novel treatment development itself. In this article, I review several major new findings relevant to our current understanding of brain alterations in mood disorders including gene-environment interactions, structural and functional brain imaging, and neurochemistry. I discuss current subtyping of mood disorders with a focus on depression severity, symptom clusters, and biology and treatment response, in suggesting the existence of endophenotypes. I also address certain major controversies in the field, including use of SSRIs in children and adolescents and whether SSRIs increase suicidality in certain patient populations. Finally, I describe new data on nonpharmacological treatments.
DEPRESSION AND HEART DISEASE
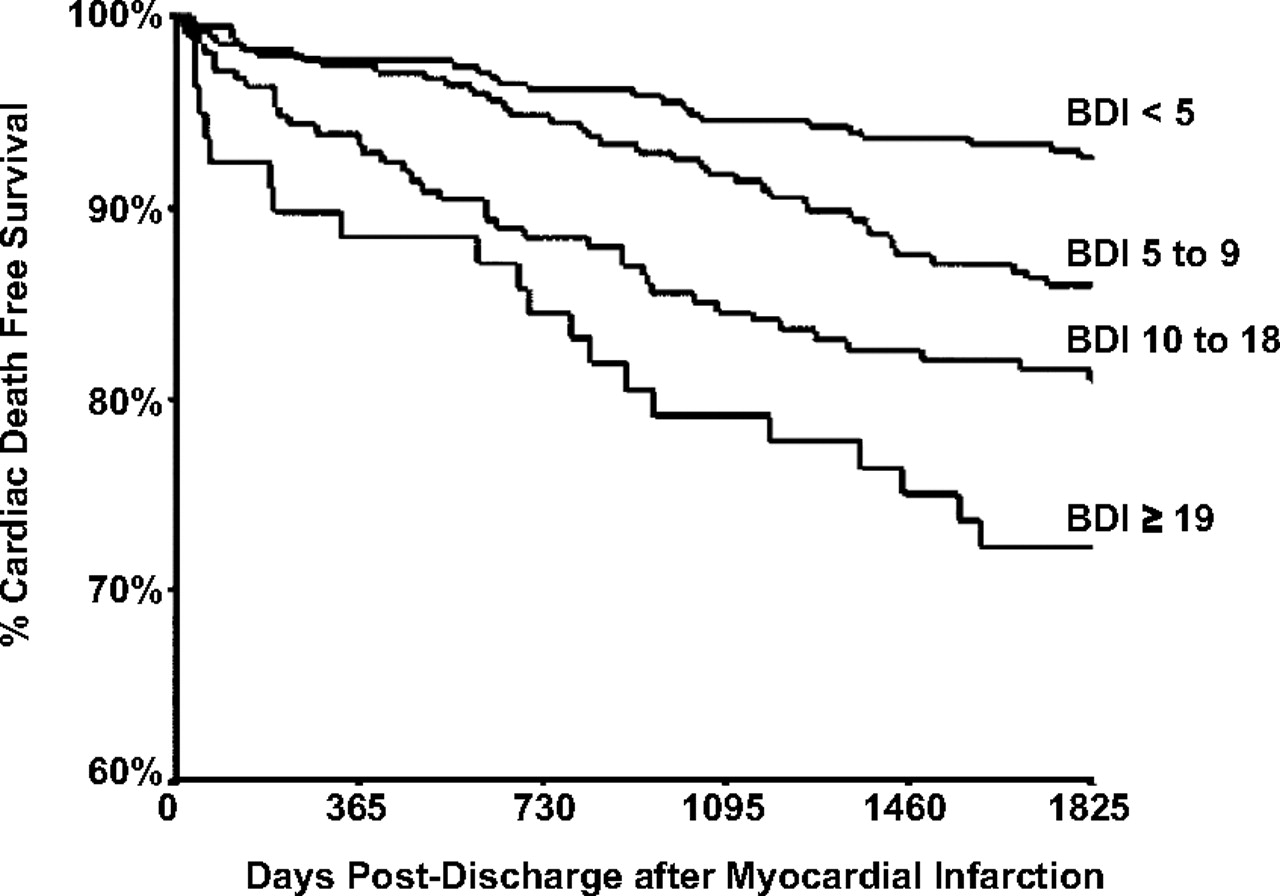
In the last two decades, a remarkable relationship has emerged between depression and cardiovascular disease. Although several medical disorders have unusually high rates of comorbid syndromal depression, the relationship between depression and heart disease has been studied much more thoroughly. In addition to the high prevalence rate of depression in patients with coronary artery disease, there is the now well-documented finding that patients with comorbid depression and various cardiovascular disorders have a much poorer outcome than those without comorbid depression. Thus, patients with comorbid depression after a myocardial infarction are more likely to die of cardiac causes than similar patients without depression, and, moreover, the severity of depression is inversely related to long-term survival (
Figure 3). This relationship has also been observed in patients after coronary artery bypass graft surgery and in patients with isolated systolic hypertension.
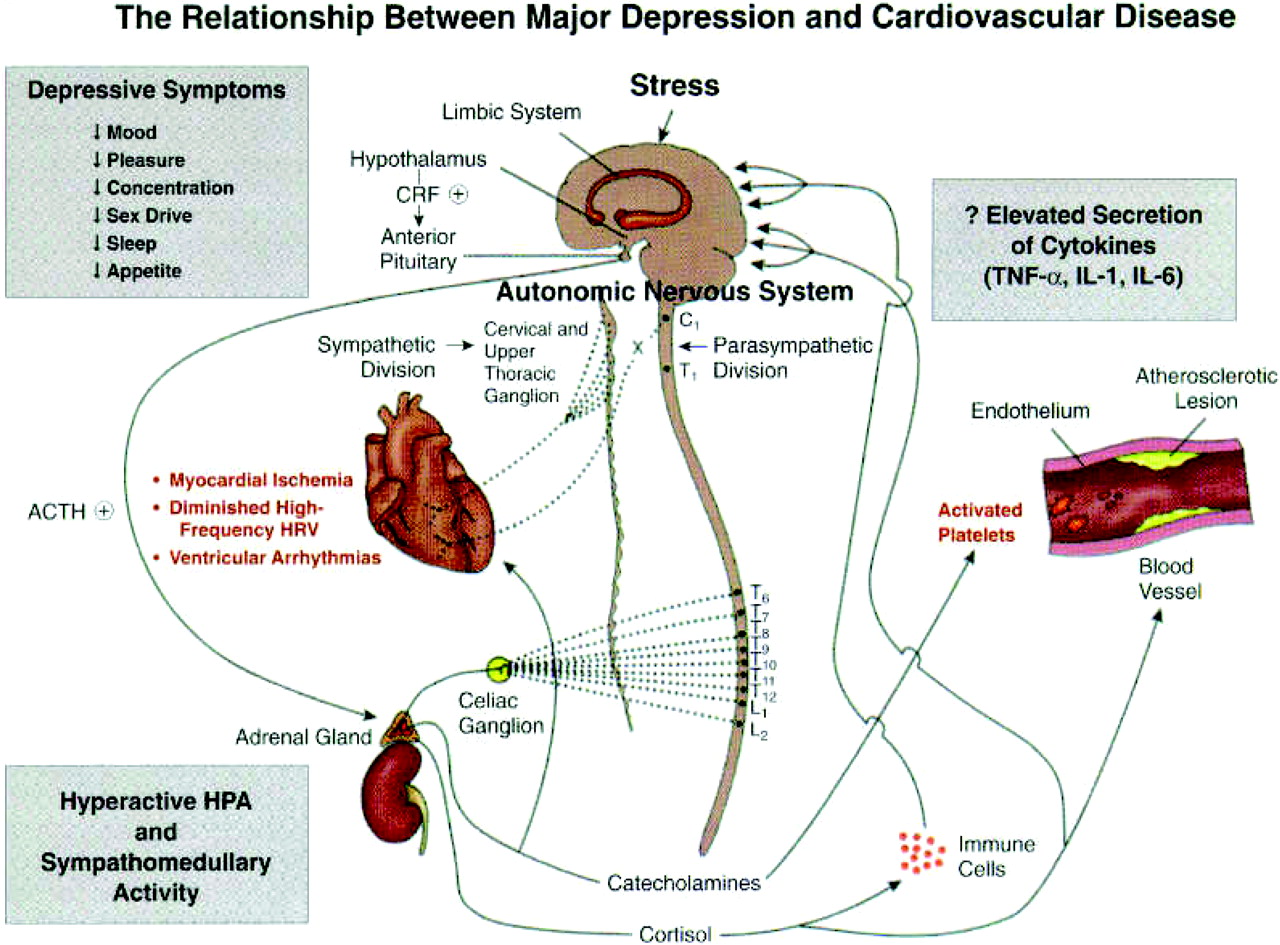
Perhaps even more extraordinary has been the repeated observation that depression is an independent risk factor for the development of ischemic heart disease—indeed, as important a risk factor as elevated plasma lipids or cigarette smoking. Research to determine the mechanisms by which depression increases both the development of coronary artery disease and the poor outcome in patients with cardiovascular disease has led to the following findings (
Figure 4).
1) Depressed patients have multiple defects in the platelet clotting cascade, resulting in a net increase in the likelihood of thrombus formation, the proximal event in both myocardial infarction and stroke.
2) Inflammation has long been known to play a role in the pathophysiology of cardiovascular disease, and there is now considerable evidence that patients with depression exhibit an increase in several markers of inflammation including inflammatory cytokines such as interleukin 6 and tumor necrosis factor.
3) Heart rate variability (HRV) is a well-validated measure of the heart's ability to respond to physiological demand. Reduced HRV is a well-established risk factor for myocardial infarction. Several studies have revealed that depressed patients without heart disease exhibit reductions in HRV. Of particular interest is the finding that treatment of depressed patients with SSRIs normalizes both their platelet clotting and HRV alterations, effects likely to also reduce the risk of myocardial infarction.
PATHOPHYSIOLOGY OF DEPRESSION
The current view of the etiology of depression is best summarized as virtually a prototype gene-environment interaction model for complex diseases such as cancer, hypertension, and diabetes, with much focus on the three major monoamine systems—serotonin (5-hydroxytryptamine, 5HT), norepinephrine (NE), and dopamine (DA). The emerging new tools of molecular neurobiology and functional brain imaging have provided additional support for the involvement of these three systems. In contrast with previous reviews (
5,
6), considerably more evidence now supports a preeminent role for central nervous system (CNS) DA circuits (
7) with many investigators suggesting that the now well-documented suboptimal therapeutic responses to SSRIs and selective serotonin-norepinephrine reuptake inhibitors (SNRIs) may be due, in part, to their relative lack of effect on brain DA circuits. As regards CNS 5HT systems, even more evidence has accrued to support a preeminent role for their involvement in depression. In addition to the very impressive evidence of reduced activity of serotonergic neurons in depression as assessed in postmortem, CSF, and neuroendocrine studies, there are new data from both postmortem and positron emission tomography (PET) imaging studies demonstrating a reduction in the number of serotonin transporter (SERT) binding sites (the site of action of SSRIs) in the midbrain and amygdala of drug-free depressed patients, as well as a reduction in both presynaptic (in the midbrain) and postsynaptic (in the mesiotemporal cortex) 5HT
1A receptor density (
8,
9). Taken together, these data suggest a net reduction in the number and/or function of the presynaptic 5HT nerve terminals and a reduction in postsynaptic serotonergic signal transduction, at least at one of the 5HT receptor subtypes. Previous studies demonstrated an increase in 5HT
2 receptor density, perhaps due to a relative decrease in 5HT availability.
Two new and important observations concerning 5HT circuits in depression are worth noting. The first is the now well replicated observation that depressed patients in remission after treatment with SSRIs, when challenged by an experimental maneuver that reduces CNS 5HT availability, i.e., tryptophan depletion, exhibit a rapid and profound return of depressive symptoms, in a few hours in many cases (
10). This finding suggests that in vulnerable individuals, reduced 5HT availability is associated with the rapid emergence of depression.
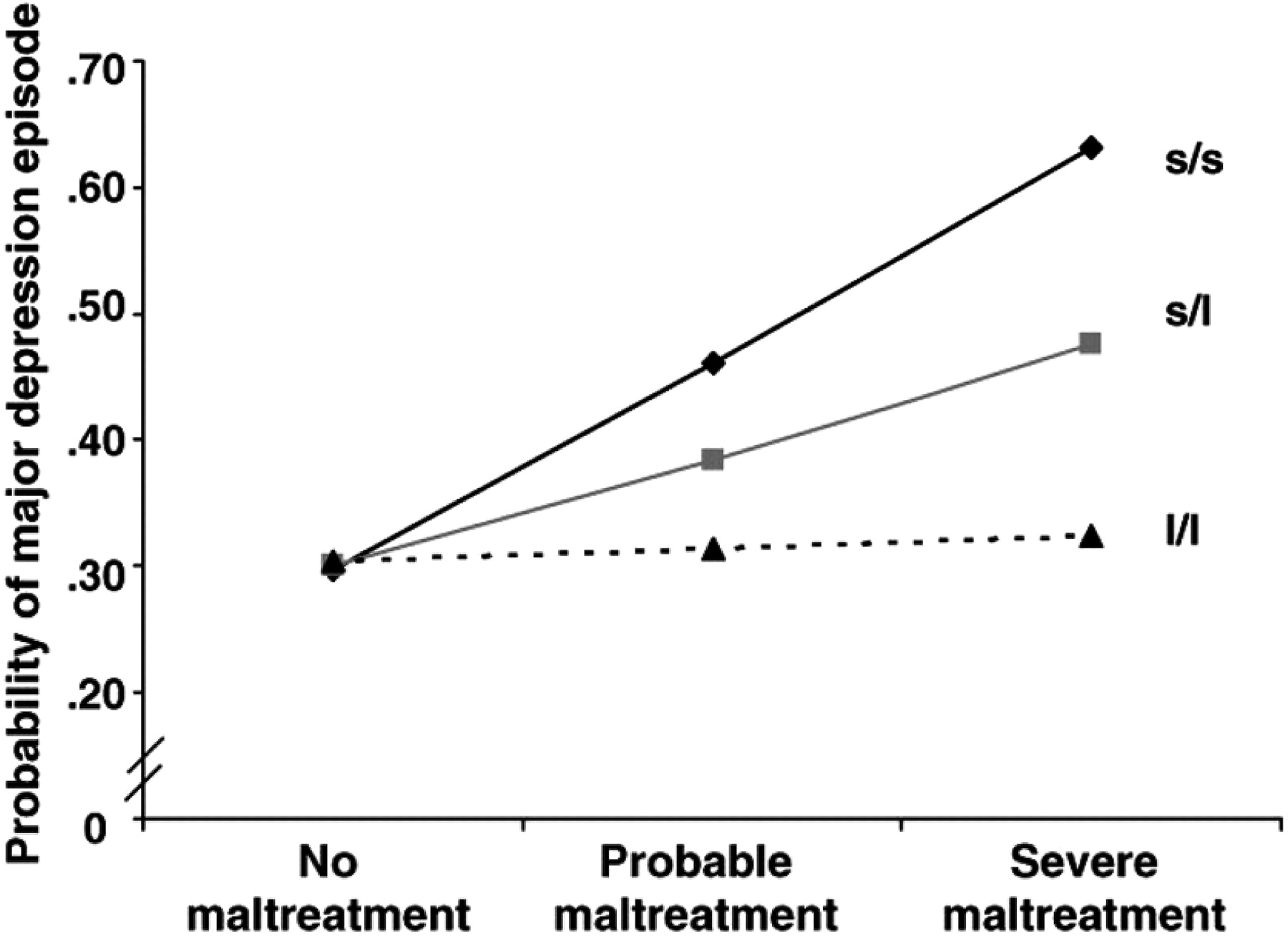
The second and, arguably, one of the most remarkable observations in all of biomedical research in the last decade is the observation that individuals with the s allele of the promoter region of the SERT gene (SLC 6A4) are unusually vulnerable to the now well-documented depressogenic effects of early life stress, i.e., child abuse or neglect, and, moreover, that this effect is “dose dependent,” both in terms of the s allele (one copy or two) and in terms of the frequency and severity of the abuse (
11). Thus, the most vulnerable to depression are individuals with the s/s genotype and the least vulnerable are those with the l/l genotype, with s/l individuals having intermediate risk (
Figure 5). This finding is all the more extraordinary because this polymorphism has been shown to be functional: s/s and s/l individuals exhibit reduced SERT binding sites in PET imaging studies compared with l/l individuals. Note that individuals with the l/l genotype are immune to the depressogenic effects of early life trauma, representing a disease-resistant haplotype. This original observation by Caspi et al. (
11) has now been replicated by the vast majority of, but not all, subsequent studies, and these data have recently been reviewed. Another allele, the Lg or Goldman polymorphism, which is similar functionally to the s allele, has recently been discovered and may account for some of the variance in the studies to date.
Table 1 summarizes the evidence showing that alterations in 5HT systems occur in depression.
Of the major catecholamine systems, NE-containing circuits have long been considered to be pathologically involved in the etiology of mood disorders (
12). Similar to drugs that increase 5HT availability, NE reuptake inhibitors such as reboxetine and nortriptyline are effective antidepressants. Moreover, neurochemical and neuroendocrine studies in depressed patients and postmortem findings support a role for NE dysfunction in depression; these data are summarized in
Table 2. Alterations in noradrenergic circuits may play a preeminent role in patients with treatment-resistant depression. Whether antidepressants that are believed to act upon both 5HT and NE neurons are more effective than those that act solely on 5HT or NE neurons remains an area of controversy, but recent meta-analyses suggest that if any advantage exists, it is quite meager (
13).
In the last decade, partly because of the disappointingly low remission rates in the large-scale clinical trials with SSRIs and SNRIs as noted above, a potential role for one or another CNS DA circuit in depression has been postulated. This emergence of a DA hypothesis of depression is not surprising in view of the fact that the inability to experience pleasure, anhedonia, is considered by many to be the most important pathognomonic symptom of depression, and pleasure, whether associated with eating, social, or sexual behavior, is well documented to be primarily mediated by DA neurons.
Table 3 summarizes the burgeoning evidence for a role for altered dopaminergic circuits in depression. In brief, both postmortem tissue and PET imaging studies have revealed reduced DA transporter binding sites (
14) and increased postsynaptic DA D2/D3 receptor density, indicative of a reduction in the synaptic availability of DA in depression. These findings suggest that treatments that enhance DA neurotransmission such as monoamine oxidase inhibitors (MAOIs), DA receptor agonists, or triple (5HT, NE, and DA) reuptake inhibitors (currently under development) may represent a novel approach to SSRI nonresponders.
STRESS, THE HYPOTHALAMIC-PITUITARY-ADRENAL AXIS, AND CORTICOTROPIN-RELEASING FACTOR
Reports indicating that a significant number of patients with depression hypersecrete cortisol, the major adrenocortical stress hormone, first appeared fifty years ago (
15). The observation that patients with Cushing's disease or syndrome often experience severe depression and anxiety and the increased production and secretion of glucocorticoids in healthy individuals exposed to stress, in part, contributed to the modern stress-diathesis hypothesis of depression. Thus, excess secretion of cortisol and other hormones of the hypothalamic-pituitary-adrenal (HPA) axis have been posited to play a significant role in the etiology of depression.
A variety of methods are now available to measure the activity of the HPA axis. The hierarchical organization of the HPA axis is shown in
Figure 6.
One of the first tests of HPA axis function to be studied in psychiatric patients was the dexamethasone suppression test (DST), a test originally designed to aid in the diagnosis of Cushing's syndrome. Small (1 mg) doses of the synthetic glucocorticoid, dexamethasone, are administered orally at 11:00 p.m., and plasma cortisol concentrations are measured at two or three time points on the following day (
16). Dexamethasone acts primarily on the anterior pituitary corticotrophs to reduce the secretion of adrenocorticotropic hormone (ACTH), resulting in a decrease in the synthesis and release of cortisol from the adrenal cortex. Failure to suppress plasma cortisol concentrations after dexamethasone administration suggests impaired feedback regulation and hyperactivity of the HPA axis.
A large percentage of drug-free patients with depression exhibit failure to suppress secretion of cortisol after administration of dexamethasone, commonly referred to as dexamethasone nonsuppression, and this was proposed as a biological diagnostic test for depression. However, multiple comprehensive analyses revealed that although many patients with depression did exhibit evidence of enhanced HPA axis hyperactivity, patients with other psychiatric diagnoses often did as well, including those with eating disorders, Alzheimer's disease, bipolar disorder, and others. However, in depressed patients, DST nonsuppression has generally been found to be associated with depression severity, and when persistent, with a significant-risk for relapse. Undoubtedly, the greatest contribution of the DST was to serve as an impetus for subsequent studies exploring the pathophysiology of the HPA axis in depression.
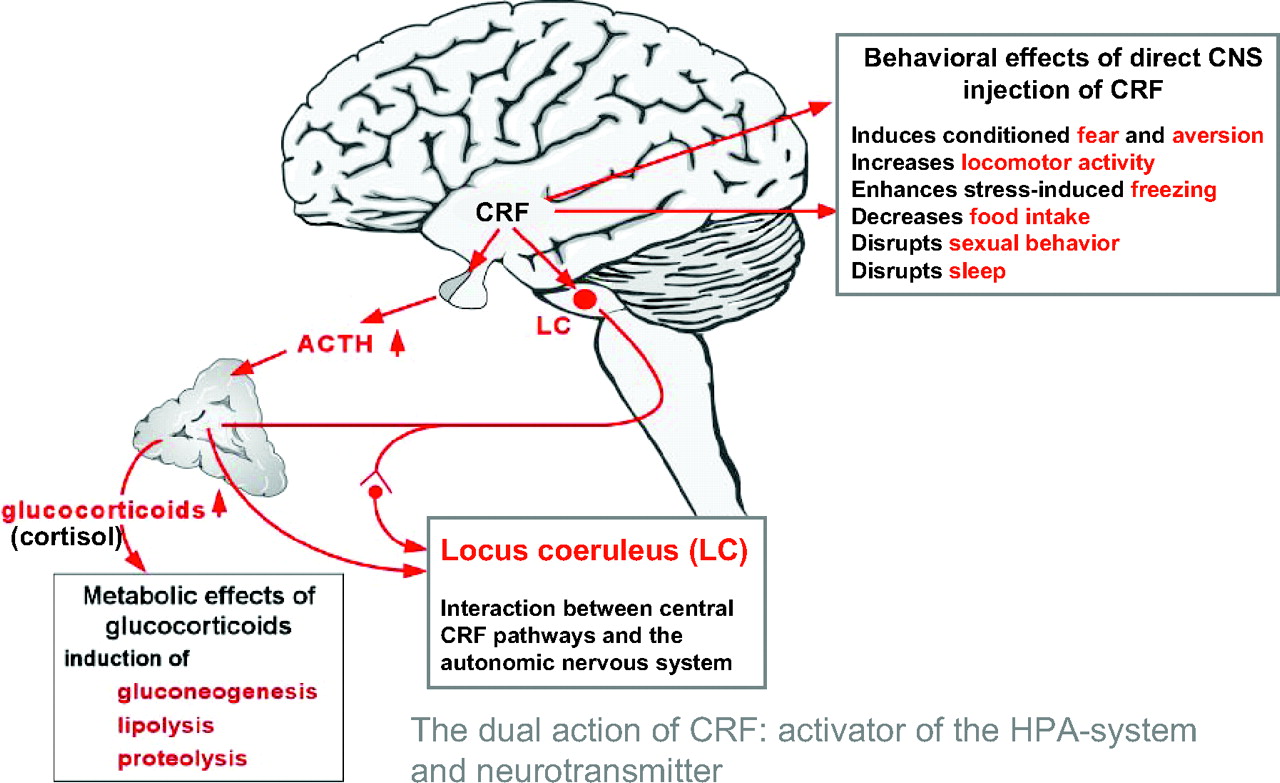
In 1981, the long sought after hypothalamic releasing hormone, corticotropin-releasing factor (CRF), a 41-amino acid-containing peptide, was discovered by Vale et al. (
17) at the Salk Institute. This singular finding greatly accelerated research on the HPA axis, stress, and depression. Neurons of the paraventricular nuclei of the hypothalamus project to the median eminence where they secrete CRF into the hypothalamo-hypophyseal portal system. CRF is then transported in this specialized vascular system to the anterior pituitary where it acts on corticotrophs to increase ACTH secretion, thereby controlling HPA axis activity. Of paramount importance was the discovery that CRF is also widely distributed in extrahypothalamic brain areas where it functions, in concert with the hypothalamic CRF system, as a neurotransmitter coordinating the behavioral, autonomic, endocrine, and immune responses to stress (
Figure 6).
The availability of synthetic CRF allowed the development and standardization of a CRF stimulation test. In this test, ovine or human CRF is administered intravenously and plasma ACTH and cortisol concentrations are measured at 30-minute intervals over a 2- to 3-hour period. Normal healthy volunteer subjects respond to CRF infusion with increased secretion of ACTH and cortisol, whereas depressed patients exhibit a blunted ACTH but normal cortisol response. Not surprisingly, the blunted ACTH response to CRF occurs in depressed DST nonsuppressors, but not in depressed patients with normal DST suppression.
Holsboer and colleagues at the Max Planck Institute in Munich, Germany (
18), developed arguably the most sensitive test of HPA axis activity, the so-called Dex-CRF test, which combines the DST and the CRF stimulation test. Thus, patients are pretreated with oral dexamethasone (1 mg) at 11:00 p.m. and are given a 100-μg infusion of human CRF on the following day. In this paradigm, depressed patients exhibit enhanced secretion of ACTH and cortisol compared with normal volunteers, indicative of HPA axis hyperactivity. Interestingly, in this test, asymptomatic first-degree relatives of depressed patients exhibit increased HPA axis activity that is maintained over time, suggesting the presence of a heritable vulnerability to HPA axis dysregulation.
Most, if not all, of the HPA axis alterations in depressed patients may be a result of chronic CRF hypersecretion. Consistent with this hypothesis, depressed patients have repeatedly been found to exhibit elevated CSF CRF concentrations (
19). Further, postmortem studies of individuals depressed at the time of death or those who committed suicide have revealed a decreased density of CRF receptors in the frontal cortex, decreased expression of CRF
1 receptor mRNA and increased CRF concentrations in a variety of cerebrocortical brain areas and the locus coeruleus compared with control subjects (
20). Successful treatment of depression with either electroconvulsive therapy (ECT) or fluoxetine, an SSRI, has been shown to result in a reduction in the high pretreatment CSF CRF concentrations (
21). Moreover, similar to continued DST nonsuppression, persistently elevated CSF CRF in symptomatically improved depressed patients is associated with early relapse of depression. These data are even more impressive when considered together with the many laboratory animal studies showing that when administered directly into the CNS, CRF produces many of the signs and symptoms of depression including decreased appetite and weight loss, decreased sexual behavior, disrupted sleep, and altered psychomotor activity (
22).
As noted earlier and illustrated in
Figure 2, approximately 30%–40% of the risk of development of depression is believed to be heritable, with the remaining variability imparted by the environment. Exposure to stress, a process primarily regulated by CRF-containing neural circuits and the HPA axis, is known to precipitate depression in vulnerable individuals. Moreover, early life stress, such as child abuse, occurring during neurobiologically vulnerable periods of development, is one of the major means whereby the environment influences the development of depression. Our group has demonstrated that depressed women with a history of prepubertal sexual abuse exhibit persistently increased HPA axis activity as evidenced by a blunted ACTH response to CRF infusion and both hypercortisolemia and increased ACTH secretion in response to a standardized laboratory stressor (
23). Using the Dex-CRF test, we recently demonstrated increased HPA axis activity in adult men with major depression and a history of child abuse (
24). Furthermore, in an inner city highly traumatized sample of patients recruited in the waiting room of a public hospital-based medical clinic, we identified individual single nucleotide polymorphisms and a common haplotype of the CRF
1 receptor that modulates the depressogenic effects of child abuse and neglect (
25). The CRF
1 receptor antagonist R121919 showed promise in the treatment of depression but was subsequently withdrawn from clinical trials because of hepatotoxicity. Other CRF
1 receptor antagonists, repeatedly shown to possess antidepressant and anxiolytic properties in laboratory animals, are novel antidepressant and anxiolytic drug candidates currently being studied in randomized, controlled, double-blind clinical trials for efficacy in the treatment of depression and anxiety disorders (
26).
OTHER NEUROTRANSMITTER SYSTEMS
Space constraints preclude a comprehensive discussion of the putative role of other neurotransmitter systems in the pathophysiology of depression. There is some evidence for the involvement of glutamate, γ-aminobutyric acid, substance P, brain-derived neurotrophic factor, thyrotropin-releasing hormone, somatostatin, leptin, and acetylcholine-containing neurons in the pathogenesis of depression (
27).
THE NEUROANATOMY OF DEPRESSION
Although there is little doubt that various neurotransmitter systems are pathologically involved in the etiology of depression, no single neurotransmitter systems appears to be solely responsible. This is not surprising when one considers the panoply of symptoms (
Figure 1) that comprise the depressive syndrome including depressed mood, loss of interest in usual activities, inability to experience pleasure, impaired concentration, disturbed sleep, decreased appetite, and suicidality. A more recent conceptual approach to the biology of depression is to consider it a systems-level disorder involving several critical brain regions and pathways involving these regions. Advances in brain imaging have allowed for rapid advances in these research areas. Structural brain imaging using magnetic resonance imaging (MRI) has generated a number of reports of altered volumes of several brain regions in patients with depression, most notably a reduction in hippocampal and caudate nucleus size and an increase in pituitary volume. As noted above, it is now evident that some of the previously described changes in certain brain structures may be more likely caused by early life stress during a critical period in brain development than to depression per se (
28). Structural brain imaging studies have also led to the emergence of a biologically distinct subtype of depression, vascular depression, first described independently by Alexopoulos at Cornell University and Krishnan at Duke University, associated with white matter hyperintensities on MRI scans and late life onset of the depressive syndrome (
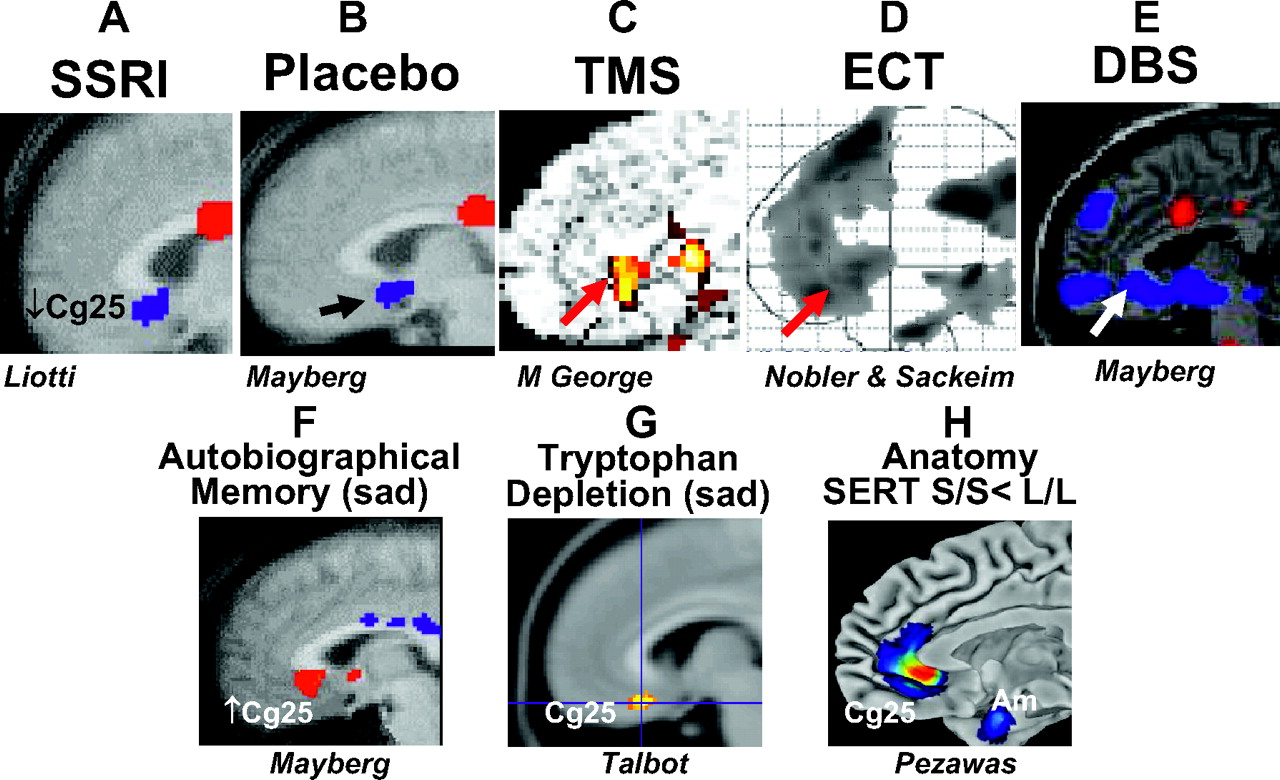
29). These morphometric studies, although revealing, have been largely supplanted by functional brain imaging studies, more specifically functional MRI, most recently with diffusion tensor imaging, as well as PET. These studies have shed light on both the involvement of specific brain regions in depression and the effects of treatment, both psychotherapeutic and psychopharmacological. PET studies led by Mayberg et al. at Emory University (
30) and by Drevets at NIMH (
31) have repeatedly supported the hypothesis that depression is essentially characterized by abnormalities in limbic system-cerebrocortical circuits, more specifically reduced activity in frontal cortical areas and hyperactivity in the amygdala and other limbic sites (
Figure 7). Most important is the repeated observation of the importance of the subgenual cingulate cortex (Cg25); this brain area shows a striking decrease in activity in response to clinical improvement of depression after treatment with SSRIs, ECT, and other novel treatments.
ANTIDEPRESSANTS AND SUICIDALITY
The question of whether antidepressants paradoxically increase the risk of suicidal ideation, suicide attempts, or suicide and if so, in which patient populations remains an active avenue of investigation and controversy. Twenty years ago, a report appeared, suggesting that an SSRI, fluoxetine, increased suicidality in a small series of adult patients with major depression. Since that time, a controversy regarding this finding has raged and has resulted in several Food and Drug Administration (FDA) hearings. The placement of a “black box” warning in package inserts of antidepressants focused on children and adolescents in October 2004 and a second warning expanded to adults in July 2005 resulted, not surprisingly, in a corresponding decline in the prescribing of antidepressants. Many studies has been conducted and several publications have appeared with no, as yet, clearly definitive answer to this critical question. Considerable attention has focused on children and adolescents, because in controlled clinical trials, a very small but significant risk for an increase in suicidal ideation and attempts was observed in association with SSRI and SNRI treatment. It is important to note that there were no suicides in any of these clinical trials. In several studies, a very strong statistically significant relationship was found between the consistent decline in the suicide rate in the United States and the rate of prescription of antidepressants, both in the nation as a whole and by region. Recent evidence, both in the United States and in other countries, has revealed a reduction in SSRI prescribing rates in response to concerns about suicide and a corresponding increase in suicide rates for the first time in many years. The available data suggest that depression is a disorder associated with a very high risk for suicide and, moreover, that antidepressants have reduced this risk. The reports of increased suicidality in some youngsters treated with antidepressants may be due to “switching” of patients with previously undiagnosed bipolar disorder from a depression to a mixed state of mania and depression. Future research should focus on identifying genetic, biological, demographic and psychological factors that predict increased suicidality in a small percentage of children and adolescents after treatment with antidepressants.
ADVANCES IN THE TREATMENT OF DEPRESSION
As remarkable as the new information on the pathogenesis of depression that has accrued over the last decade has been, even more impressive is our increased understanding of the current state of the treatment of this disorder.
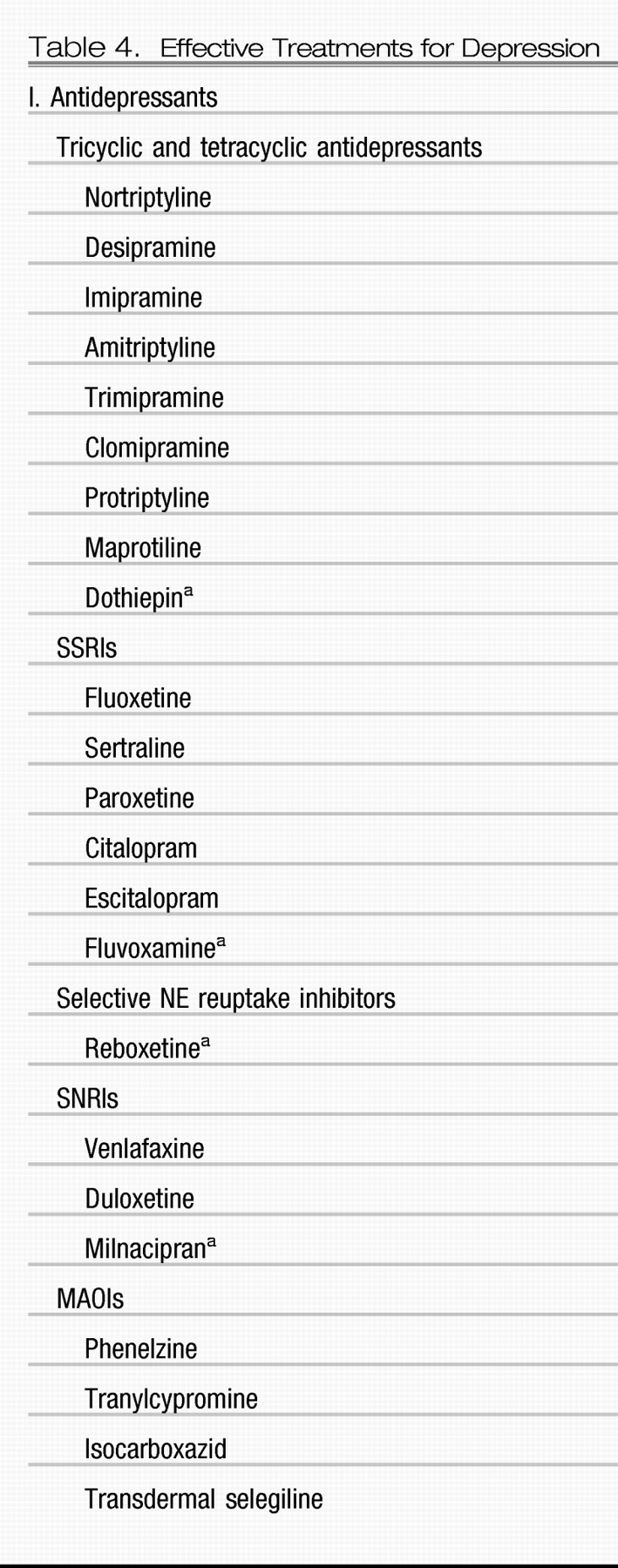
Table 4 lists the currently approved and investigational treatments for depression. As noted earlier, the results of a large multicenter National Institutes of Health (NIH) sponsored effectiveness study, STAR*D, and multiple placebo-controlled, randomized, double-blind clinical trials have revealed a disappointing rate of remission (in the range of approximately 28%–45%) in acute monotherapy trials with SSRIs and SNRIs (
3). These results indicate that more than half of depressed patients exhibit either no benefit or only a partial benefit after an adequate trial with the most commonly prescribed agents. The good news is, however, that we not only now know a great deal more about the use of augmentation and combination strategies, but we have also witnessed the development of novel treatments including vagus nerve stimulation and deep brain stimulation. In the 41-center, NIH-sponsored STAR*D effectiveness study, treatment of 2,876 outpatients with major depression with the SSRI citalopram for up to 14 weeks led to a remission rate of only 28%, using the Hamilton Depression Rating Scale as the outcome measure. The patients who did not attain remission with citalopram monotherapy were either switched to be treated with bupropion (unknown mechanism of action, N = 239), sertraline (an SSRI, N = 238), venlafaxine (an SNRI, N = 250), or continued citalopram combined with either bupropion (N = 565) or buspirone (a partial 5HT
1A agonist, N = 286). In the switch arm, patients who did not achieve remission with citalopram and were treated with bupropion exhibited a 21.3% remission rate, those treated with sertraline exhibited a 17.6% remission rate, and those treated with venlafaxine exhibited a 24.5% remission rate. In the combination arm of the study, patients treated with citalopram and bupropion attained a 29.7% remission rate and those treated with citalopram and buspirone attained a 25.3% remission rate. In a third level of this study, 142 patients in whom treatment in levels 1 and 2 failed, were then treated with lithium (N = 69) or thyroid hormone (N = 73), both previously shown to convert antidepressant nonresponders to responders, for up to 14 weeks. Remission rates were 15.9% with lithium augmentation and 24.7% with thyroid hormone augmentation. In the fourth level of the study, patients in whom three or more trials failed were treated with either an MAOI (tranylcypromine) or venlafaxine + mirtazapine. The former results in a remission rate of 6.9% and the latter in a remission rate of 13.7%. These data and those of many other trials have provided evidence for the use of rational combination (and augmentation) pharmacotherapy in the treatment of major depression, approaches commonly used to successfully treat cancer, hypertension, diabetes, and other complex multisystem disorders.
Another important issue in the treatment of depression is the role of psychotherapy and, in particular, which type of psychotherapy is effective. In addition, several studies have addressed the use of a combination of an antidepressant and psychotherapy. There is now good evidence from controlled studies that at least two types of psychotherapy: CBT, originally developed by Beck at the University of Pennsylvania, and interpersonal psychotherapy (IPT), largely studied at the University of Pittsburgh and Columbia University, are effective in the treatment of major depression and often, but not always, are as effective as antidepressant medications.
In a landmark study, Keller et al. (
32) studied 681 patients with a particularly virulent form of depression, chronic depression, defined as a single episode of major depression that persisted for 2 years or longer. In fact, the mean duration of the current episode of depression for these study patients was 8 years. The patients were randomly assigned to one of three treatment groups: 1) nefazodone, an antidepressant, 2) the cognitive behavioral-analysis system of psychotherapy (CBASP), a form of psychotherapy for patients with chronic depression based upon principles of CBT and IPT, or 3) nefazodone and CBASP. This study sought to determine the relative efficacy of an approved antidepressant, an effective form of psychotherapy, and the combination in a difficult-to-treat patient population. In the acute 12-week treatment phase, the remission rate with nefazodone treatment was 33%, with CBASP was 30%, and with the combination of nefazodone and CBASP was 48%. Thus, the combination was superior in efficacy than either treatment alone. Conceptually this finding fits well with the functional imaging studies of Mayberg et al. (
33) that revealed differences in the neuroanatomical substrates of antidepressants and CBT. Interestingly, when we reanalyzed the data from this large study of chronic depression, we made two novel observations. First, the rate of child abuse and neglect in the patients with chronic depression was inordinately high, i.e., 67%. Second, patients with a history of childhood trauma (sexual, emotional, or physical abuse or neglect) responded better to psychotherapy than to the antidepressant, which when taken together with the neurobiological observations noted above in patients with major depression who have a history of early life stress, further support this type of depression as a distinct subtype—with its own unique genetic risk factors, functional brain imaging and neuroendocrine alterations and treatment response.
Other advances in treatment for depression have occurred. These include approval by the FDA of a new MAOI, selegiline, administered by a transdermal system, i.e., a patch. It clearly has fewer side effects and there are fewer concerns about dietary and drug-drug interactions than with the older MAOIs, such as phenelzine or tranylcypromine.
In addition to the augmentation and combination studies cited above, there is also evidence for the efficacy of other pharmacological combinations in the management of treatment-resistant depression. These include most notably the combination of atypical antipsychotic agents such as risperidone, aripiprazole or olanzapine with SSRIs (
34) and the combination of SSRIs and mirtazapine, an antidepressant that acts primarily on 5HT and NE receptors.
Also of note is the demonstration of a rapid and relatively long-lasting effect of a single dose of ketamine, an
N-methyl-
d-aspartic acid antagonist, in the treatment of refractory depression (
35). Perhaps, most groundbreaking has been the emergence of somatic nonpharmacological treatments for the treatment of depression. Of course, ECT has long been considered to be the most effective treatment for depression. In the past decade, the FDA has approved vagus nerve stimulation, previously approved for the treatment of pharmaco-resistant epilepsy, for treatment-resistant depression. There is now considerable evidence for the efficacy of repetitive transcranial magnetic stimulation in the treatment of depression. Based on her brain imaging studies, Mayberg has pioneered the use of deep brain stimulation using an electrode aimed at the subgenual cingulate cortex (Cg25) and obtained markedly positive results in four of six patients with extremely refractory depression (
36). Schlaepfer and colleagues at the University of Bonn, Germany, have obtained promising results with deep brain stimulation in the nucleus accumbens (37), a major DA terminal area, involved in reward and pleasure, in patients with refractory depression.
The next decade should result in a series of unmet needs for diagnosis and treatment of depression finally being realized—most importantly, biological tests to predict the best treatment, pharmacological, psychotherapeutic or nonpharmacologic somatic treatment, or combination therapy for individual patients. This treatment will probably comprise a combination of genomic testing and functional brain imaging. This advance based upon elucidation of validated endophenotypes of depression will also accelerate novel treatment development. Taken together, such advances should reduce the terrible burden of depressive illness.