The 21st century holds great hope for individuals with neuropsychiatric illnesses. Scientific advances in psychopharmacology, psychosocial therapies, somatic therapies, and neuroimaging are rapidly entering the clinical arena. Valuable new insights informed, for example, by genetic inquiry and new conceptual approaches to diagnosis promise to change the care of people living with mental illness in the very near future.
ETHICAL PRINCIPLES IN HUMAN STUDIES
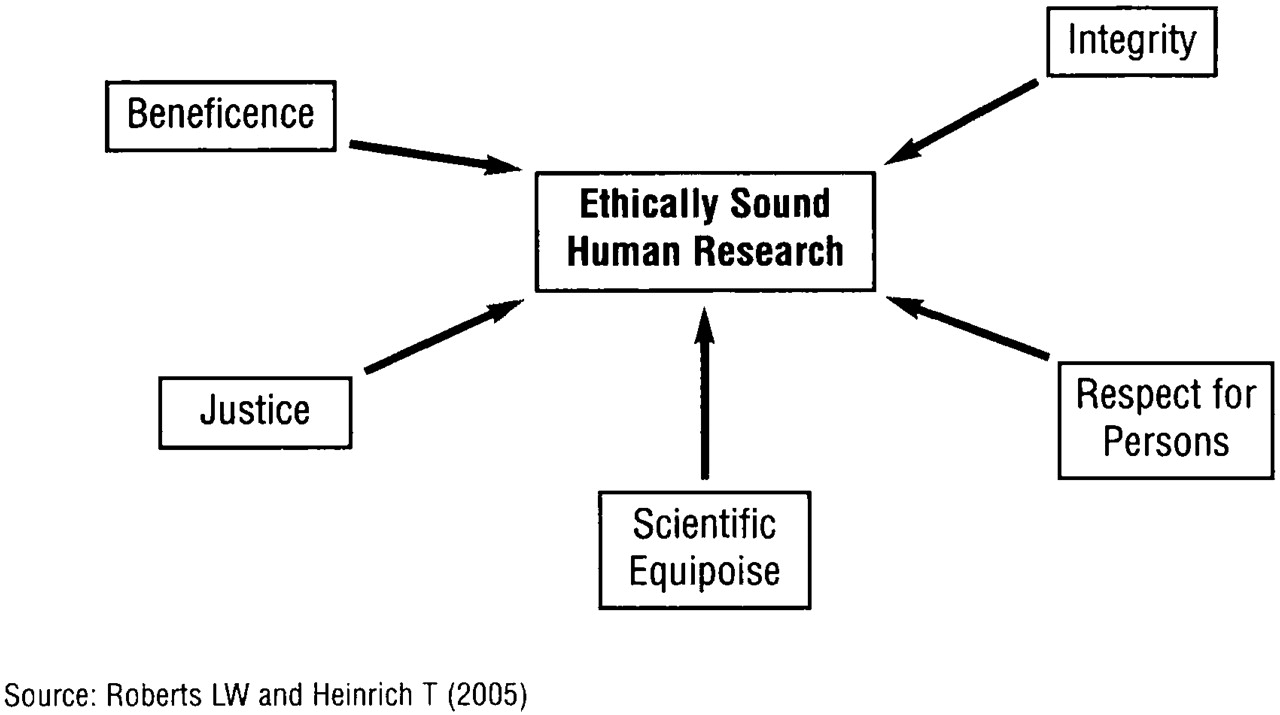
Through iterative efforts over many years, several key concepts have been defined as essential to the design and implementation of ethically sound research with human participants. The Belmont Report (
4), published by the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research in 1979, described three necessary ethical precepts:
respect for persons (dignity, autonomy, privacy, and fundamental human regard accorded to all research volunteers);
beneficence (human science should be undertaken to bring forward beneficial new knowledge through studies that maximize benefit and minimize harm); and
justice (fairness in the distribution of burdens and benefits of research and in the selection of study populations).
A fourth principle,
integrity, has emerged with equal salience (
5,
6) and refers to consistent adherence to the ideals of the professions. It is closely linked to fidelity (i.e., “faithfulness”) and veracity (i.e., truthfulness).
A fifth concept,
scientific equipoise, borrowed and reinvented for its ethical meaning from the scientific design literature, refers to the idea that the body of existing scientific knowledge is insufficient to answer a question of genuine importance (
7–
9). Therefore, a scientist is poised between two possibilities (i.e., treatment A works better than no treatment or treatment A works better than treatment B), and an experiment is needed to clarify between them. Stated differently, it is unethical to include a person in an experiment with its inherent burdens and possible risks if the scientific answer is already known.
Respect for persons, beneficence, justice, integrity, and
scientific equipoise are believed to be necessary preconditions for ethically sound research (
Figure 1), but these five commitments are not sufficient unless they are translated fully in the intentions and actions of every member of the research endeavor (
2,
10). Moreover, the expectations of researchers naturally and necessarily evolve as the shared ethical constructs of a society change and as we learn more about ethically meaningful aspects of human experience. This said, the shared understanding among bioethicists and scientists is that this suite of ethical concepts may help present-day scientific studies stand the test of time.
SAFEGUARDS IN HUMAN STUDIES
Ethical concepts find concrete expression in what are referred to as “human subject protections” or safeguards. The most well known of these safeguards are those mandated in federal regulations, such as review and oversight by institutional review boards, clinical trials registries, informed consent, data safety monitoring boards, and confidentiality measures.
Beyond these formal safeguards are many more ways to protect study volunteers across the course of research participation (
11,
12), for example:
•.
Pursuing only scientific questions that have importance and meaning
•.
Designing scientific projects that use the minimum exposure to risk necessary to answer the scientific question
•.
Studying the least vulnerable population necessary
•.
Using recruitment techniques that are neither coercive nor deceptive
•.
Endeavoring to obtain fully informed and authentic consent from volunteers
•.
Maintaining appropriate steps to ensure the privacy of participants
•.
Proactively managing any potential or actual conflicts of interest
•.
Debriefing participants at the conclusion of their involvement in a protocol
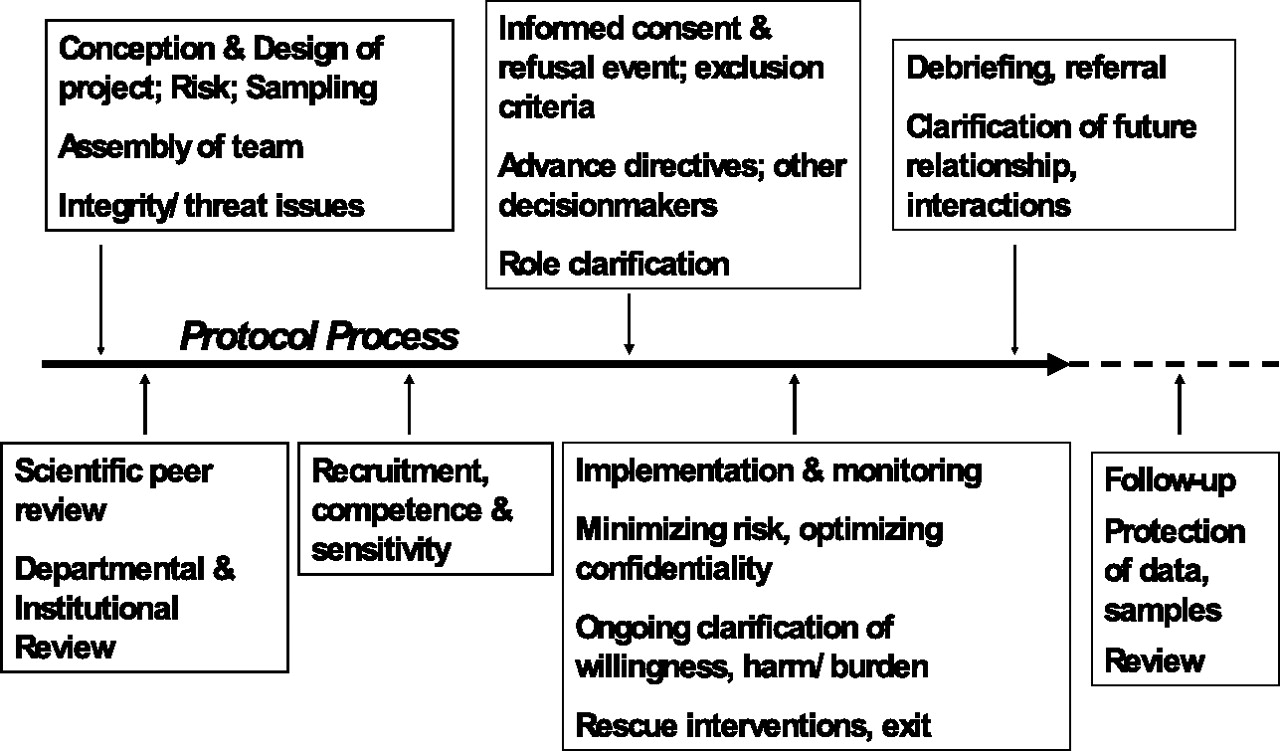
The ethical foundations of proposed research are built on the ethical awareness and strength of the research team (
Figure 2). The process of conducting ethical psychopharmacological research starts with scientific expertise and the integrity and professional standards of the investigators and stakeholder organizations (e.g., the academic medical center and the source of funding for the project). The scientific design and the project methodology should be crafted with ethical considerations in mind. For example, it is ethically problematic to place a human volunteer in a life-threatening risk situation to study an issue that itself is not life-threatening.
Criteria related to scientific design and safeguards can help researchers construct research protocols that are ethically “defensible.” These are referred to as the “necessity criteria” and are key elements in protocols that have adequate and appropriate protections for participants included in the design and implementation of the proposed research endeavor (
6,
10). The necessity criteria, for instance, may provide a general overview of how to evaluate the ethical appropriateness of psychopharmacological studies. Second, the goals of the protocol should, necessarily, be salient to the population studied. Third, the least vulnerable population should be used to study the issue in question. When more vulnerable populations are required or more dangerous protocols are used, the ethical guidelines must be monitored more rigorously.
Institutional review boards (IRBs) are involved in research oversight and monitoring of research protocols and use criteria (such as the “necessity criteria”) to evaluate the ethical soundness of research. As a study is being developed, an IRB reviews its scientific importance, research design, and subject protections according to federal guidelines and state and local policies. Once the study question has been deemed sufficient to warrant the undertaking of a research protocol, attention must be paid to developing a research methodology that minimizes the risk of potential harm to study subjects.
The issue of placebo controls in research is an example often raised as a potential source of harm to research subjects who, as part of the protocol, may be taken off previously effective treatments and given a placebo, which may lead to relapse and clinical decompensation. Placebos may be viewed as violating the Declaration of Helsinki (
13) of 1964, which asserted that subjects are owed the “best proven diagnostic and therapeutic method.” Proponents of the inclusion of placebo controls in neuropsychiatric research assert that placebo arms help detect adverse events and accurately determine clinical benefit within the active arm of the study (
14). Adequate clinical monitoring and thorough documentation coupled with an appropriate mechanism for rapid corrective intervention is necessary in the attempt to minimize risk to study subjects. Researchers also routinely include stopping rules in protocol proposals. Such rules state that when it becomes clear that one arm of the protocol is significantly superior to or, conversely, more dangerous than another arm, the study should be stopped regardless of the originally proposed study termination point.
The IRB is also often responsible for ongoing oversight of the research. For example, it may request regular progress reports and information on adverse events encountered during the course of the study. The IRB's considerations in proposed scientific undertakings that involve vulnerable populations (e.g., children) are often unique and more rigorous in an attempt to minimize the risk of exploitation and harm. At a minimum, greater attention is given to issues of risk and assent/consent processes. With genetic research in children, for example, the emerging ethical issues were previously unimagined and unimaginable and because the physical risks are far outweighed by serious but unanticipated psychosocial risks, IRBs have a very difficult responsibility.
Recruitment of volunteers into a biomedical research protocol requires an appreciation of participants' potential vulnerabilities, which may influence the decision to enter or remain in a study. For example, an economically disadvantaged person may choose to enroll because of what is perceived as a valuable compensation arrangement, or a decisionally compromised person may choose to participate because he or she does not understand how research differs from usual care. For these reasons, recruitment and informed consent processes are tightly linked and perhaps seamless from the perspective of the participant. Informed consent entails the communication of accurate, detailed, and balanced information to potential study volunteers.
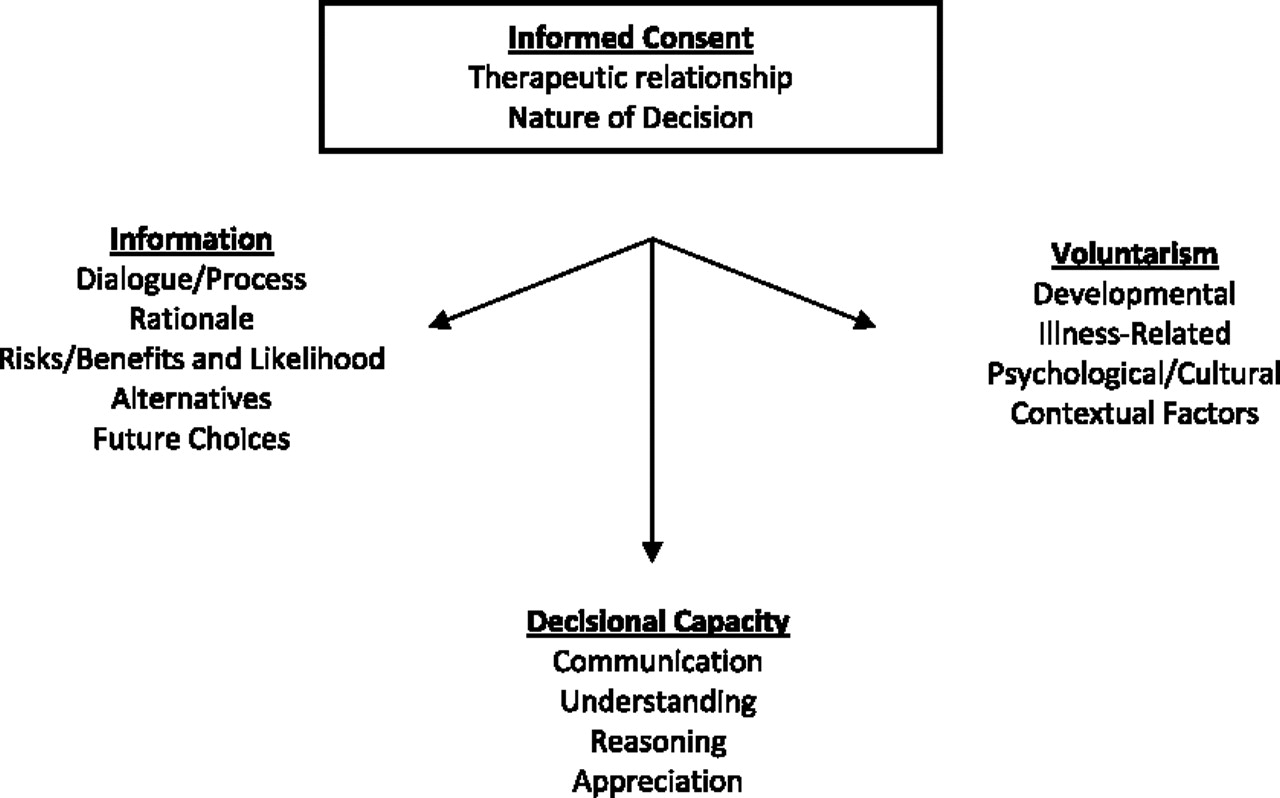
The process of providing informed consent involves ensuring that a study participant is able to engage in an independent and thoughtful decision-making process before enrollment and requires that the person have sufficient decision-making capacity to make this kind of choice (
Figure 3) (
15). The informed consent process comprises three important and unique components: disclosure of pertinent information including risks, benefits, and alternatives (as well as that of not participating in the proposed research protocol); voluntariness (a genuine choice made without coercion); and decision-making capacity (a concept with multiple attributes; see
Figure 4) (
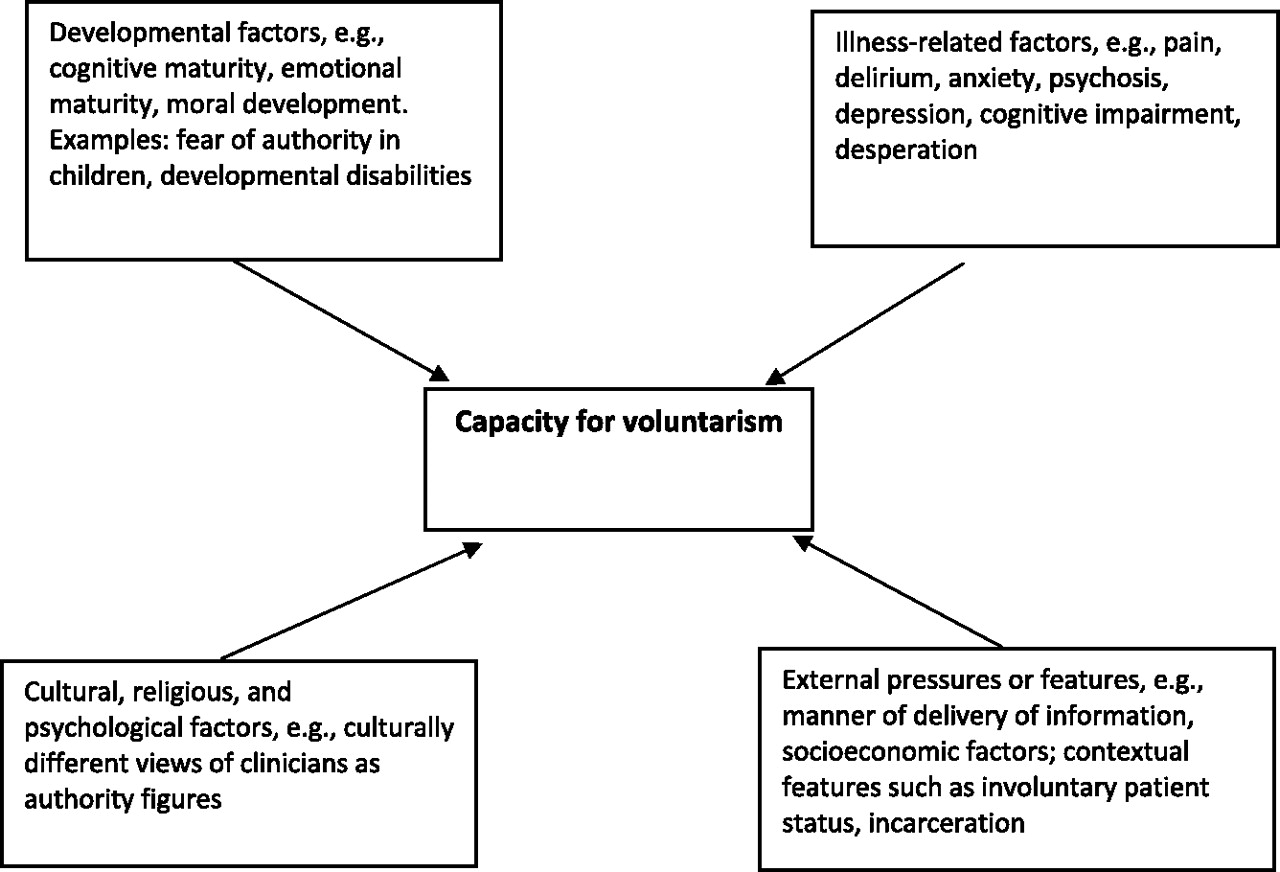
16). Being able to make an authentic, voluntary decision is also necessary and is influenced by age and developmental level, illness-related considerations (e.g., amotivation and negative cognitive distortions such as hopelessness), psychological and cultural issues (e.g., whether one feels or is empowered to make an individual decision), and contextual factors (e.g., institutionalization or poverty) (
16).
An individual's capacity to provide informed consent is not a fixed attribute but may differ depending on the level of complexity and risk involved in the study and the constraints experienced at the time of decision. Because different types of decisions require different degrees of capacity and different decision-making skills, the ability to provide consent varies from situation to situation. A participant may be able to consent to one research protocol (e.g., a low-risk study with great potential benefit) but at the same time lack the capacity to consent to another research protocol (e.g., a high-risk study with little probable benefit).
Establishing alternative decision makers or research advanced directives before the start of a study provides a potential ethical safety net for research participants who may experience symptoms or deterioration in their clinical status during a protocol. For example, research subjects may experience periods when they are unable to make decisions that guide their participation in the research protocol, and psychiatric research advance directives could help direct appropriate care regarding withdrawal from a protocol or the use of rescue medications. However, a recent review found that too few data are available to make definitive recommendations about the use of psychiatric advance directives at this time (
17).
Once the research project is underway, close monitoring of results and protocols by involved individuals and institutions is imperative. Researchers must be aware of trends appearing in preliminary data that may reveal unanticipated risks or burdens being placed on the volunteers. Criteria for the appropriate use of rescue strategies or disenrollment must be upheld. When the study is completed and results are submitted for publication, journal editors and peer referees must be diligent in their evaluation of the ethical standards of the work.
Proactively managing any potential or actual conflicts of interest is another key safeguard. The dual roles of a physician-researcher may come into conflict during a study. In the clinical setting, the physician's duty is to provide treatment with the goal of advancing the patient's well-being. In the research setting, the scientist has the obligation to generate data that attempt to adequately answer the research question posed. Maintaining this differentiation of roles requires attention and diligence. Despite the physician-researcher's best efforts, a “therapeutic misconception” may arise in which the researcher or the research subject fails to appreciate the ethical differences between clinical care and scientific research (
18–
21). This misconception of the relationship between researcher and research subject may impair the subject's ability to provide a fully informed and authentic consent.
Research collaborations with the pharmaceutical industry can offer valuable opportunities but must be carefully balanced against the risk of significant conflicts of interest (perceived or real). For example, a recent, well-publicized review of 74 clinical trials of antidepressants revealed that 37 of 38 studies with positive results were published, but only 3 of 36 studies with negative results were accurately represented in the literature (
22). Data from multiple studies show that a majority of potential research subjects want to know about researchers' related financial relationships (
23). It is therefore important that the investigator disclose any potential financial conflicts, but ideally these potential conflicts of interest would be avoided entirely by the research team.
Beyond these efforts, it is also critical to study the least vulnerable population necessary to answer the scientific question at hand. National guidelines (e.g.,
http://www.hhs.gov/ohrp) outline specific steps to be taken to protect particularly vulnerable populations such as children, pregnant women, and elderly individuals. However, individuals with various neuropsychiatric illnesses may also be considered vulnerable, particularly when they are institutionalized or have other societal disadvantages such as poverty. They may exhibit impaired decision-making abilities, making obtaining adequate and authentic informed consent a challenge. However, deficits in understanding (or other domains of capacity) do not necessarily justify excluding someone from participation in research. Rather, such deficits indicate the need for enhanced methods of consent, such as by providing consent information in an interactive and iterative format that facilitates sharing and, for many, may improve decisional abilities (
24).
It is important to note that relatively few safeguards in human research have been formalized in federal regulations or policies. Most, in fact, are completely in the hands of researchers and affirmed through professional ethics standards. Training in ethics is crucial for clinicians and scientists alike. Learning the ethical benchmarks of neuropsychiatric research will provide future investigators a framework for addressing the inevitable ethical quandaries raised as they develop their own body of work. Furthermore, all practicing psychiatrists should be able to critically evaluate the ethical foundation of published research. APA (
http://www.psych.org) and the American College of Neuropsychopharmacology (
http://www.acnp.org) have prepared excellent resource documents that articulate the multifaceted ethics expectations of neuropsychiatric and psychopharmacological researchers. Very thoughtful ethics documents for authors and editors have also been developed (
25).
RESOLVING ETHICAL QUESTIONS
Ethical considerations in neuropsychiatric and psychopharmacological research have gained great salience over the past two decades (
2). A majority of the most challenging ethical questions fall into three main domains. The first pertains to sources of vulnerability and informed consent-related issues, such as fluctuating or diminished decisional capacity in a person with schizoaffective disorder, inclusion of people with cognitive disorders who are institutionalized, and enrollment of children with mental illness or addiction-related conditions. The second domain relates to research design issues, such as the conditions under which placebo comparisons are needed in randomized, controlled studies and the ethical use of symptom-provocation or state-altering maneuvers in neuroimaging studies (
26–
28). The third domain relates to overlapping role issues in psychopharmacology research and the interactions of clinicians with the pharmaceutical industry.
New and very difficult issues have arisen for all areas in biomedical research, expanding well beyond the scope of psychiatry and encompassing considerations such as integrity/conflict of interest issues, the adequacy of the premarket testing of medications, and Health Information Portability and Accountability Act (HIPAA)-related confidentiality protections in human research (
29,
30). The data ownership, publication right requirements, and financial incentives built routinely into contracts for some industry-based clinical trials, for instance, have raised concerns about whether these projects can fulfill the positive duties of beneficence and integrity. Adverse effects of medications that are unexpectedly more prevalent or more severe when introduced to broader patient populations and used over time have raised questions about the adequacy of U.S. Food and Drug Administration oversight and current practices in protecting public health interests. The boundaries and practices associated with identifiable data in research projects have been revised, and specific issues related to privacy and confidentiality will increase dramatically as genetic and psychopharmacogenetic studies become commonplace (
31).
Despite what seems to be an exponential growth in ethical questions and concerns, there is also a sense of growing optimism that we are developing better—more sensitive, more refined, and more effective—approaches to ethical challenges inherent to human studies. Taken together, criteria and evidence pertaining to scientific and ethical issues constitute invaluable tools for resolving ethical dilemmas. Furthermore, these tools can be used prospectively for identifying and preventing ethical problems and enhancing the ethical attunement and skills of investigators and reviewers.
Criteria related to scientific design and safeguards, as mentioned previously, can help researchers construct ethically “defensible” projects with appropriate protections for participants (
6,
10). Careful scientific and ethical criteria may also help dismantle ethical questions accompanying the use of placebo comparison arms in protocols (
28). The concepts of beneficence and justice suggest that research involving a treatable condition uses a comparison that is at least equal to usual standards of care. Although the balance of scientific evidence on this point is changing, many believe that the significant placebo response of some people with depression suggests that placebo comparison can, under some circumstances, be justified in testing the effects of antidepressants and certain psychotherapies for depression, thereby meeting the scientific equipoise criterion. Evidence of the effectiveness and efficacy in antipsychotic medications makes this a harder argument, yet expectations for federal agencies such as the Food and Drug Administration may still include placebo comparisons as the gold standard for approval of new agents. Nonetheless, investigators involved in studies using placebos have a great ethical responsibility for ensuring that appropriate safeguards are in place if, for instance, while a participant is taking a placebo, he or she develops suicidal thoughts or requires rescue medications.
An emerging field of evidence-based ethics (
32,
33) has also given rise to data of immense value in resolving ethical dilemmas. Whether study volunteers are uncertain of the roles, duties, and role tensions for clinical investigators and whether they understand the differences between clinical research and clinical care (i.e., the “therapeutic misconception”) are questions for which the answers are becoming clearer through studies of perspectives and understanding of actual clinical research participants (
18,
19,
34). These findings can help investigators and institutions develop approaches (e.g., separation of roles and provision of additional information) that eliminate misunderstandings and misplaced expectations. Similarly, studies of different information-sharing, decision-coaching, and voluntarism-enhancing procedures and of the strengths and deficits of research volunteers have begun to shed light on ways to improve protocol safeguards (
32).