PRESENTATION OF THE CASE
A 78-year-old right-handed physician presented to the Neurological Clinics of the University of California, Los Angeles, with a 6-month history of progressive “confusion” and gait difficulty. His family first noted periods of disorientation and lack of clarity and coherence in his thinking. They became alarmed when the patient mixed up the identities of his daughters and began having difficulty grasping concepts and remembering instructions and directions. Shortly after the onset of his symptoms, the patient underwent an elective knee replacement for degenerative arthritis. Postoperatively, he showed significant neurologic deterioration from which he never recovered. His mentation became slower and he was less able to handle complex tasks. Moreover, after the surgery he started to complain of numbness and tingling in both feet.
Over the next few months, the patient had a nonfluctuating, gradually deteriorating course. Although he continued to work as a practicing physician, he was reportedly falling asleep while seeing patients. He was no longer able to drive long distances and was getting lost in unfamiliar surroundings. His lower extremity paresthesias progressed up his legs to the top of his calves and interfered with his gait. He now walked with an extremely cautious gait and had sustained several falls. Despite these symptoms, he continued to make medical decisions and to handle his instrumental activities of daily living. The patient's past medical history was otherwise negative for medical illnesses including strokes, hypertension, cardiovascular disease, diabetes, alcoholism, or toxic exposures. He did not take prescription medications or recreational drugs. His family history included a mother who had slowly progressive dementia in her eighties. On review of symptoms, the patient endorsed a decline in his mental sharpness but denied other cognitive symptoms (such as memory difficulty or word-finding problems) or psychiatric symptoms (such as depression or hallucinations).
His mental status examination revealed a patient with psychomotoric slowing and decreased spontaneity of speech and behavior. His family did most of the talking for him; he initiated little conversation, but responded to questions. There was a noticeable latency in his responses, and his answers were short and unelaborated. When left unstimulated, the patient fell asleep at several points during the interview. On the Mini-Mental State Examination,
1 he had a score of 27 out of 30, missing 1 point each on orientation, registration, and his copy of the intersecting pentagons. Despite his slowness, his digit span was 7 forward and 4 backward, and he could do continuous performance tasks and sustain attention. The patient's language examination revealed normal levels of fluency, auditory comprehension for simple commands, confrontational naming, repetition, reading, and writing. He had difficulty with multiple-step items but could do the component steps without difficulty. The patient generated a word list of 17 animals in a minute. On an auditory verbal learning test, the patient had mild difficulty with delayed recall but responded well to cueing and recognition. In contrast, he was unable to correctly copy a three-dimensional cube and other complex drawings. There were no problems in performing learned motor movements or double-digit calculations, and he demonstrated normal abstraction to similarities, idioms, and proverbs. The patient was able to complete Luria alternate programming and motor sequencing tasks without evidence of perseveration. There was no evidence of special preoccupations or other neuropsychiatric conditions.
Vital signs and physical examination were normal except for a surgical scar on his left knee. There was no evidence of meningismus. On neurological examination, his cranial nerves were intact. Gait was hesitant, slightly broad-based, and unsteady. The Romberg test was negative, and there was no dysmetria or disdiadochokinesia. The motor evaluation showed normal strength and tone. There were no fasciculations, but the patient had mild upper-extremity action tremors. His deep tendon reflexes were symmetrical and 1+ in the upper extremities but absent at the knees and ankles. There were no pathological reflexes and toes were downgoing. On sensory examination, he had decreased touch and pinprick as well as vibratory sensation in a stocking distribution in the lower extremities symmetrically to just below the knees.
The patient underwent an extensive evaluation. All routine laboratory results were normal, including blood cell counts, metabolic panel, and thyroid, renal, and hepatic functions. Antinuclear antibodies, rheumatoid factor, and syphilis serology all were negative, and protein electrophoresis was normal. Chest X-ray was unremarkable. His electroencephalogram (EEG) showed mild generalized symmetrical slowing. Magnetic resonance imaging (MRI) of the brain showed mild nonspecific cerebral atrophy and only very mild periventricular white matter changes on T2-weighted images. Nerve conduction study and electromyography of the lower extremities were consistent with a distal, symmetrical, demyelinating sensorimotor polyneuropathy.
The patient's subsequent course was rapidly progressive. He developed increased confusion, somnolence, visual hallucinations, and frequent falls. During one fall he broke his arm. He developed spontaneous (non-startle) myoclonus and paratonic rigidity, and his neuropathy progressed to involve his hands. Six weeks after his initial evaluation, a second EEG showed significant generalized slowing without periodic waves or epileptiform activity. A gadolinium-enhanced brain MRI showed mild atrophy and no enhancing lesions, and brain single-photon emission computed tomography (SPECT) showed widespread decreased perfusion involving posterior parietal areas, both hippocampal areas, the anterior cingulate gyrus, and the central white matter. Laboratory tests for inflammatory, infectious, and paraneoplastic processes were negative, including for anti-Hu and anti-Yo antibodies. Cerebrospinal fluid (CSF) evaluation showed normal cell counts, protein, glucose, and syphilis serology. All CSF cultures were negative. CSF immunoglobulin levels were normal, and there were no oligoclonal bands. In addition, the patient's CSF when evaluated at the National Institutes of Health was negative for protein 14-3-3.
2The clinical course continued to be one of relentless deterioration. The patient was admitted to a nursing home, where eventually he became mute and showed little spontaneous behavior. In addition, he developed hyperorality, putting non-food items in his mouth. The patient developed aspiration pneumonia and died 9 months after his initial presentation.
CLINICAL DISCUSSION
The patient exhibited cognitive deficits consistent with a frontal-subcortical dementia. He demonstrated slowed psychomotor activity, problems with multistep commands, and decreased spontaneity and behavioral initiation. He had a mild memory retrieval difficulty, abnormal constructions, and a lack of insight into his illness. The added decreased alertness with somnolence if underaroused suggested delirium; however, the patient did not have an acute confusional state with prominent attentional deficits and daily fluctuations. His behavioral syndrome was consistent with criteria for dementia, especially if the memory retrieval difficulty was considered sufficiently severe.
3,4The pattern of deficits was more consistent with frontal-subcortical dementias than with Alzheimer's disease (AD) or other cortical dementias. Psychomotor slowing, behavioral aspontaneity, and memory retrieval difficulty indicate dementing illnesses that affect frontal-subcortical circuits.
3 In particular, the aspontaneity points to disturbance of frontal systems involved in motor initiation. Moreover, this patient lacked the cognitive deficits characteristic of AD, such as true amnesia, prominent language difficulty, and ideomotor apraxia. His executive abilities were difficult to assess; however, his lack of insight into his disease suggested related frontal-executive dysfunction. His late hyperorality further suggested an element of the Klüver-Bucy syndrome from progression to anterior temporal areas.
5The patient's disorder had a rapid course to death within 15 months of onset. The initial evaluation eliminated most rapidly progressive frontal-subcortical dementias from consideration. These disorders include toxic-metabolic disturbances; subacute CNS infections including AIDS, syphilis, fungal or viral meningoencephalitides, and Whipple's disease; and CNS neoplasms such as gliomatosis cerebri and angiotropic lymphoma.
3 The initial history, examination, and neuroimaging failed to disclose evidence for these conditions. In addition, the patient's blood and CSF results eliminated most metabolic or infectious processes. The clinicians strongly considered the possibility of a paraneoplastic process from an occult malignancy. Paraneoplastic limbic encephalitis, however, tends to present acutely with a pure amnestic syndrome and increased T
2-weighted signal of the mesial temporal lobes on MRI. Tumors such as small-cell carcinomas of the lung may be associated with confusion, seizures, and the presence of circulating paraneoplastic, usually anti-Hu, antibodies. This patient did not have lesions on MRI or evidence of circulating antibodies associated with paraneoplastic syndromes.
The rapid progression also suggested the diagnosis of Creutzfeldt-Jakob disease (CJD). This disorder is a rare fatal disease that usually includes a rapidly progressive frontal-subcortical dementia, startle myoclonus, gait difficulty, and periodic (1–1.5 cps) biphasic and triphasic waves on EEG.
6 Patients with CJD may also have rigidity (extrapyramidal, paratonic, or both), primitive reflexes, brisk deep-tendon reflexes, cerebellar dysfunction, and occasional cortical blindness and progressive aphasia. Most patients with CJD have a subacute course leading to death within months (median of 4 months, mean of 5–8 months); however, 8% to 9% percent have a more chronic course, usually of 2 to 4 years.
6,7 CJD is usually sporadic, nonfamilial, and due to abnormal conformation of the prion protein (PrP), an unconventional infectious pathogen with an incubation period of months to decades. CJD is potentially transmissible, and universal precautions are indicated in the handling of blood, spinal fluid, and potentially contaminated instruments from patients suspected of this disorder. Given his family history of dementia, however, familial transmission was a consideration in this patient. Other related prion diseases that show familial transmission are Gerstmann-Sträussler-Scheinker disease (GSSD) and fatal familial insomnia (FFI).
8,9 The patient was less likely to have these other prion disorders than CJD because of the absence of ataxia and serious insomnia.
The clinical diagnosis of CJD is often difficult. In the absence of a reliable clinical test or specific neuroimaging changes, establishment of the diagnosis of CJD requires pathological demonstration of a spongiform encephalopathy.
6 During life, the most characteristic clinical features of CJD are myoclonus and periodic EEG sharp wave complexes, but both of these can be absent, and myoclonus can occur in AD and other dementias.
10 Our patient failed to demonstrate periodic sharp waves on EEGs, but he developed myoclonus later in his course. Structural neuroimaging in CJD is usually normal or demonstrates only atrophy or nonspecific signal abnormalities in the basal ganglia on T
2-weighted MRI images. Recently, hyperintensity in the basal ganglia and in the cerebral cortex on diffusion-weighted MRI (DWI) has been reported.
11,12 Functional neuroimaging with positron emission tomography or with SPECT usually demonstrates changes consistent with diffuse areas of hypometabolism. Results of routine CSF analysis are usually normal or demonstrate mildly elevated protein levels. Some investigators have found an abnormally high level of CSF neuron-specific enolase in CJD.
13 Recently, investigators reported that elevated CSF levels of the 14-3-3-brain protein are highly sensitive and specific for CJD.
2,14–16 In our patient, the 14-3-3 protein was not detected in the CSF.
The patient's dementia was associated with development of a peripheral neuropathy. Early in his course, this patient developed progressive paresthesias in the lower extremities, which progressed to a severe distal, symmetrical sensory polyneuropathy with gait disturbance. A search for the etiology of peripheral neuropathy was unrevealing. Nevertheless, the occurrence of dementia with peripheral neuropathy narrows the differential diagnosis (
Table 1). Dementia plus a distal symmetrical neuropathy most commonly represents a toxic-metabolic disorder or a rare inherited disorder, particularly if the pattern is that of a symmetrical distal sensorimotor neuropathy. Another strong consideration is adult polyglucosan body disease, a disorder of polysaccharide metabolism associated with a frontally predominant dementia with sensorimotor peripheral neuropathy and gait disturbance.
17 Adult polyglucosan body disease and most other dementing disorders associated with a distal symmetrical neuropathy have an axonal neuropathy, not a demyelinating one. In our patient, electrophysiological study and slowed nerve conduction velocities confirmed the presence of a predominantly demyelinating polyneuropathy. CJD is one of the few dementing illnesses associated with a demyelinating neuropathy. Although prion diseases are essentially limited to the CNS, investigators have reported an association of peripheral demyelination in patients with CJD associated with the E200K mutation.
18–20 The pathogenesis of this neuropathy is unclear because investigators did not demonstrate deposition of the PrP in the peripheral nerves.
When the clinical picture in this case is compared with recent classifications of sporadic CJD,
21 our patient would fulfill criteria for clinically possible CJD (
Table 2). He had a rapidly progressive frontal-subcortical dementia with eventual myoclonus and akinetic mutism. He did not meet proposed criteria for probable CJD because of the absence of the typical periodic complexes on EEG or the presence of the 14-3-3 protein in the CSF. The added presence of a demyelinating peripheral neuropathy, however, is not considered in these criteria. The demyelinating neuropathy increases the clinical diagnostic likelihood for CJD as the final diagnosis in this patient.
PATHOLOGICAL DISCUSSION
Brain-only autopsy was performed. External examination revealed mild symmetrical frontal lobe atrophy. Microscopic sections from multiple regions of the brain showed an unusual combination of neuropathologic changes. Severe spongiform change, consistent with that seen in CJD, was noted,
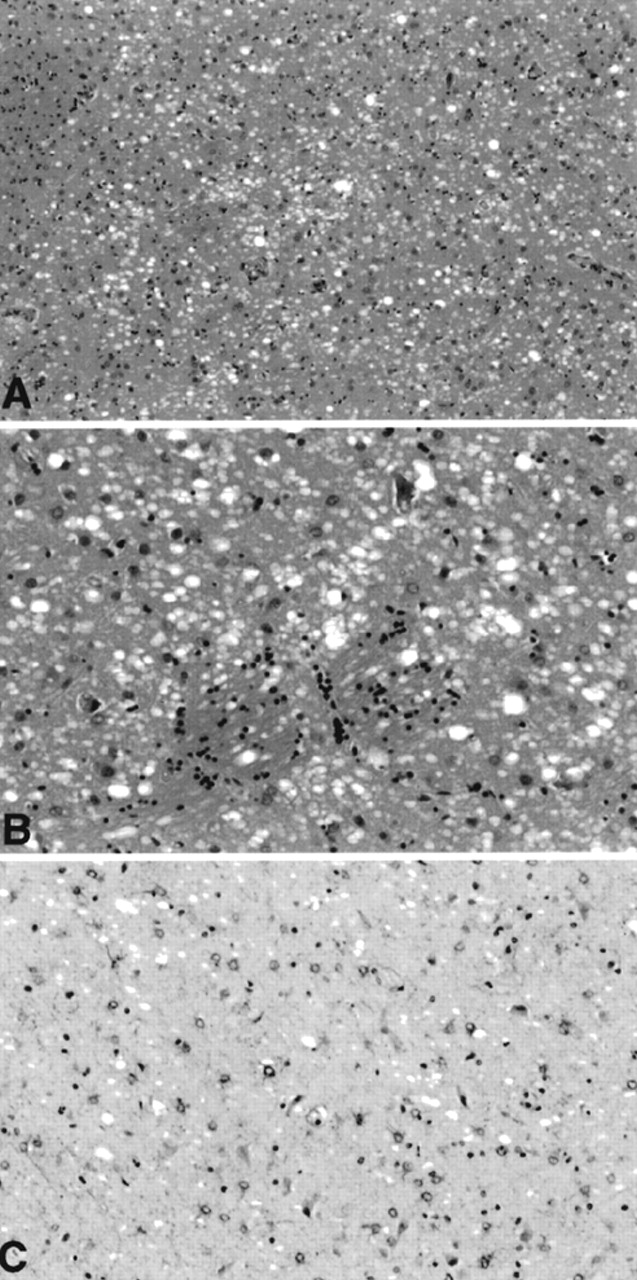
22,23 although in a somewhat unusual distribution. Prominent spongiform change was seen in the basal ganglia, including the caudate nucleus and putamen, and the thalamus, with associated astrocytic gliosis best demonstrated on glial fibrillary acidic protein (GFAP) immunohistochemistry (
Figure 1). Sections of neocortex showed surprisingly inconspicuous spongiform change, but sections of the hippocampi demonstrated severe microvacuolization. This was most prominent in the prosubiculum and subiculum, although extending throughout the pyramidal cell layer, and was accompanied in some regions by prominent astrocytic gliosis. In the hippocampal pyramidal cell layer, many neuropil vacuoles appeared to originate from neuronal cell bodies. Where seen in neocortex, the spongy change tended to be most prominent in the deep cortex.
A definite diagnosis of CJD requires neuropathological confirmation. Human prion diseases display a range of histopathological phenotypes. Five basic histological lesions are observed in prion diseases: 1) spongiosis; 2) astrogliosis; 3) neuronal loss; 4) prion protein (PrP) deposits which are Congo red positive (plaques) or negative; 5) neurofibrillary tangles. The relative severity of these five individual basic lesions and their rostrocaudal distribution within the brain result in the three pathological phenotypes that are commonly distinguished in prion diseases: CJD, GSSD, and FFI. The distribution of these changes in the brain differs in the various diseases. For example, in CJD the “status spongiosus” is mainly cortical, but amyloid plaques are usually not prominent. In GSSD, degenerative changes occur prominently in the cerebellum, and amyloid plaques are abundant. However, spongy change may occasionally be seen also in other degenerative brain diseases.
In CJD, the primary neuropathological feature is spongiform degeneration associated with accumulation of immunocytochemically detectable prion protein (PrP),
24 a pathological, amyloidogenic isoform of the expressed cellular PrP.
25 Typically, spongiform change is noted in neocortical areas, thalamus, basal ganglia, and the molecular layer of the cerebellum; the hippocampus is spared.
26 However, recently Kaneko et al.
27 found neuronal loss and astrogliosis in the parasubiculum and the external principal lamina of the presubiculum and suggested that these structures were vulnerable to early lesions in the parahippocampal gyrus in CJD.
The neuropathological hallmark of FFI (familial and sporadic fatal insomnia) is loss of neurons and astrogliosis in the thalamus independently of disease duration.
8,9,28,29 The mediodorsal and anterior ventral thalamic nuclei are invariably and severely affected; the involvement of other thalamic nuclei varies. The inferior olivary nuclei also show neuronal loss and gliosis in most cases. In contrast, the pathology of the cerebral cortex varies in proportion to the disease duration and is more severe in the limbic lobe than in the neocortex. The entorhinal cortex and, to a lesser extent, the piriform and paraolfactory cortices show spongiosis and astrogliosis in most subjects. The neocortex is spared in subjects with a disease duration of less than one year, it is focally affected by spongiosis and gliosis in those with a course between 12 and 20 months, and it is diffusely involved only in subjects with a disease duration longer than 20 months. In addition, the frontal, temporal, and parietal lobes are affected more severely than the occipital lobe. Other structures are virtually normal or show mild focal pathology.
In GSSD, histopathologic changes are characterized by presence of Congo red–positive amyloid plaques composed of PrP fragments. The amyloid plaques can coexist with significant spongiform degeneration, neurofibrillary degeneration, or amyloid angiopathy.
30To summarize, the spongiform changes observed in the brain of this patient were consistent with CJD but in an unusual distribution affecting predominantly ganglionic and diencephalic structures, suggesting the “diencephalic” variant of CJD.
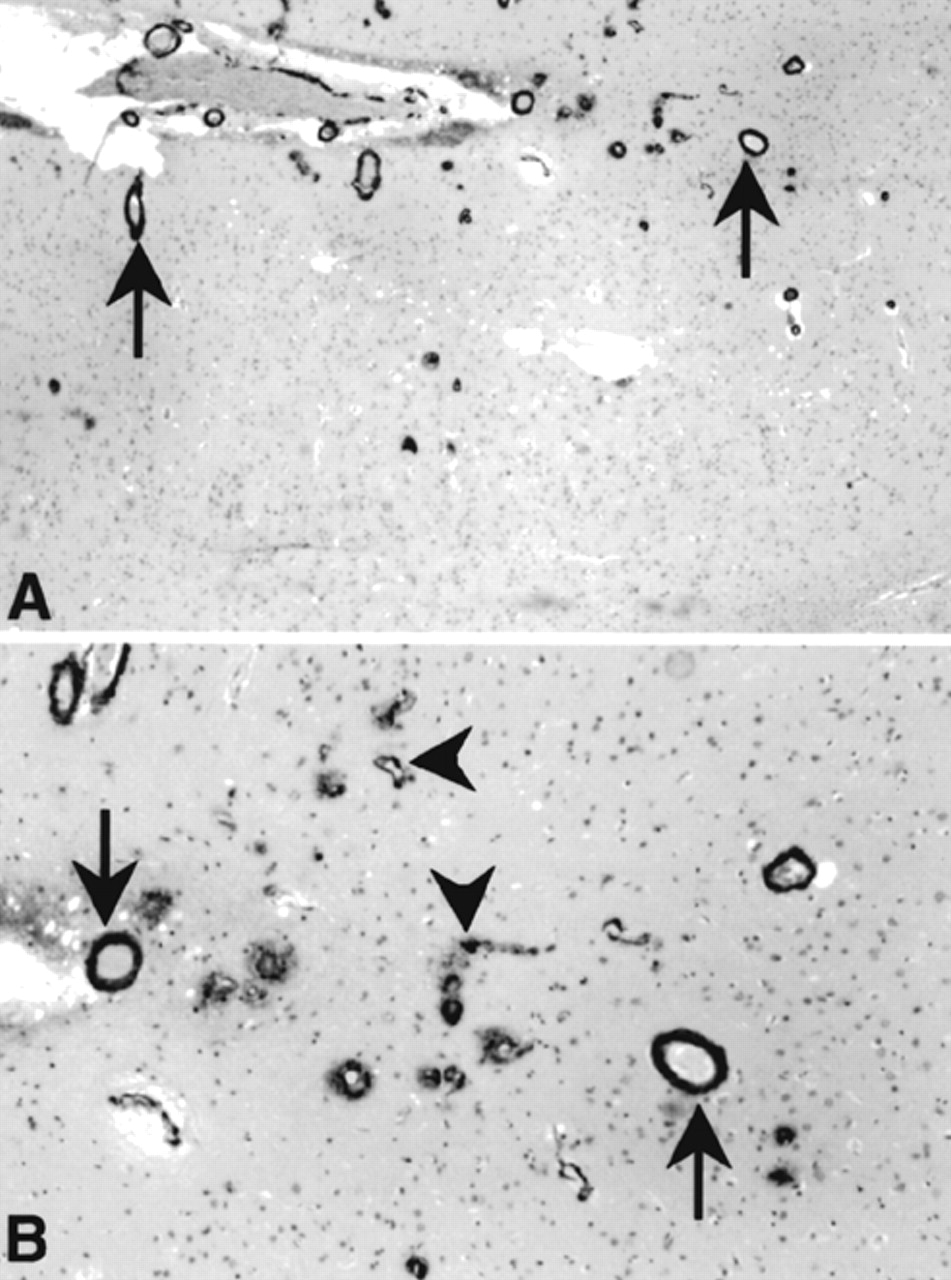
The second major neuropathological change noted in this patient was the presence throughout neocortex of severe cerebral amyloid angiopathy (CAA), affecting both arterioles and capillaries (
Figure 2). This was very widespread, involving arterioles in all regions of the cortex. Amyloid infiltrating arteriolar walls was strongly immunoreactive with antibodies to Aβ.
31 Although Aβ CAA is one feature seen in patients with AD, other histopathologic changes in this patient's brain would not meet either the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) or Braak and Braak criteria for AD;
32,33 that is, the hippocampi show only sparse neurofibrillary tangles on silver stains, at most rising to the level of “moderate,” which is inconsistent with an AD diagnosis.
31–33 The CAA was of such severity that in one or two foci, evidence of secondary associated fibrosis of the vessel walls, with early microaneurysm formation, was noted. Cerebellum showed subtle microvacuolization of the molecular layer with relative preservation of the Purkinje and granule cell layers.
Few studies on single patients have reported the coexistence of CJD and AD.
34,35 More systematic review of the coexistence of CJD and AD by Hainfellner et al.
36 revealed that changes as in definite and probable CERAD AD occur in almost 11% of CJD patients and in an even higher frequency in elderly nondemented control subjects.
36 These findings suggest that a coexistence of Alzheimer-type pathology in CJD most likely represent an age-related change. In one study, accumulation of deposits of prion protein was noted at the periphery of Aβ immunoreactive plaques.
Association of PrP amyloid and beta/A4 amyloid deposition in the same brain has occasionally been described in elderly patients with GSSD or with CJD.
37,38 Rare cases of CJD and CAA in the absence of Alzheimer changes have been described.
39–41