Both autism and familial mood disorder give evidence of strong heritability, and both are subjects of vigorous genetic investigation. In this review I develop a hypothesis that a subgroup of idiopathic autism constitutes a phenotype of familial mood disorder with prominent developmental and cognitive dimensions. This is an attempt to “broaden the biological concept,” as we repeatedly learn must be done to encompass new genetic knowledge. Examples immediately at hand include the wide range of phenotypic expressions of the genes for Rett syndrome, tuberous sclerosis, and myotonic dystrophy, among many others.
The objective of this review is to marshal and evaluate the clinical and biological evidence at every level pertaining to a possible etiological relationship between autism and familial mood disorder. Finally, the answer must rest on genetic studies. (Note that for simplicity autism will be used to designate the subgroup associated with familial mood disorder, and includes autistic spectrum disorder when so associated. The term familial mood disorder includes major mood disorder (bipolar disorder (I and II), major depression, and schizoaffective disorder); obsessive-compulsive disorder and anxiety disorder were sought, but the data concerning them are less reliable.) The areas to be discussed include the following:
I. Clinical Observations
II. Family History
III. Pharmacologic Response
IV. Neuroimaging Studies
V. Neuropsychological Observations
VI. Hemispheric Lateralization of Function
VII. Neuropathology
VIII. Neurochemical Studies
IX. Genetics
I. Clinical Observations
Early Observations of Autism and Major Mood Disorder
Any fundamental understanding of childhood neuropsychiatric disorders arguably requires that they be put in register with the categories of adult psychopathology. This idea was vindicated, I believed, by recognition (by myself and others) of childhood bipolar disorder. But that was not what led me to relate autism to major psychopathology. I initially searched for the cause of autism by traditional neurological localization. Both pathology and neuropsychology suggested that bilateral medial temporal lobe disease, or even extensive left temporal lobe disease, could produce a clinical picture simulating autism.
1However useful this idea might be as a model, it did not account for the bulk of cases of autism. Not only did most autistic children have no demonstrable lesions in the temporal lobes by available techniques, but true idiopathic autism was much more variegated and interesting than the bilateral temporal lobe model. Some of the children with idiopathic autism developed sophisticated if bizarre language, and some became competent or even precocious in one or other area of mental function (including reading or extraordinary memory feats). Some had overwhelming mood storms, and some had normal or even superior intelligence by standard testing. The sheer range of functional capacity within the group of autistic children seemed to preclude any simple formulation invoking brain damage.
At this point we noticed that an impressive number of the parents of our autistic patients had histories of manic depression. Undoubtedly we were attuned to this by our concomitant exploration of childhood manic-depression. Soon we could show, to our satisfaction, that the incidence of manic depression and major depression was significantly higher in the families of our autistic patients than in published data for the general population. A related finding reported that socially phobic, hermitic men were common in the families of patients with Asperger's syndrome.
2 We also looked more carefully at the bipolar (switching to that terminology) children we were identifying. Many of them had unusual or partial intellectual gifts or fixated obsessive interests.
3 Likewise, some of the autistic children were recognized to have cyclic disorders of mood with extreme mood disturbances, sometimes responsive to lithium. In short, we became familiar with a consistent semeiological interplay between the two seemingly disparate entities.
4 This was repeatedly reinforced by the family pedigrees.
Incidence of Major Mood Disorder in Families of Autistic Children
Other investigators have likewise found a robust elevation in incidence of major mood disorder in families of autistic children.
5–8 The interpretations of this observation have been puzzling. Some authors have gone to great length, it seems, to avoid any implication that major mood disorder and autism might be etiologically related.
7,8 In discussing their findings, they do not mention this as a possible interpretation. Piven and Palmer noted that family members of one group of autistic individuals displayed a “broader autistic phenotype,” which did not include mood disorder.
8 I take them to mean that they found two separate groups within the autistic probands: one with family histories of major mood disorder, and one with the “broader autistic phenotype.” I am inclined to agree that idiopathic autism can be divided conveniently into two “taxa,” as some have designated.
9 One is higher functioning, often with preserved islands of skills and prominent anxiety, obsessiveness, mood disorder, positive family history of major mood disorder, and frequently a family history of unusual intellectual ability/achievement. The other is lower-functioning, with prominent language disability and family members having mild language disability, rigidity of personality, and related features suggesting the “broader autistic phenotype.” In this taxon, however, there is a lack of major mood disorder and no family history of mood disorder or unusual intellectual ability/achievement. One caveat about this formulation is that the “broader autistic phenotype” is not fully defined, and one may suspect that it could be interpreted to include withdrawal, social anxiety, obsessive features, and the like, which may just as well be interpreted as features of major mood disorder.
Studies of the Relationship of Autism and Major Mood Disorder
Recently, others have given attention to the relationships between autism and major mood disorder. Ghaziuddin and colleagues find that depression is the most common psychiatric disorder accompanying autism (in contrast to earlier observers who maintained that depression was rare in autistic patients).
10 Wozniak and colleagues, surveying 727 psychiatrically referred children, conclude that “comorbid mania among patients with PDD may be more common than previously thought”, and suggest that “identification of the comorbid condition may have important therapeutic and scientific implications”.
11 An open trial of divalproex sodium showed benefit in patients with autism spectrum disorders, including improvement in core symptoms of autism.
12 Improvement was particularly seen in those with associated features of mood instability, impulsivity, and aggression, including those without a seizure history or abnormal EEG. The implication is that the agent was acting as a mood stabilizer. We have had similar experience with lithium in children with autistic spectrum disorder.
13Comparison of Clinical Features of Autism and Major Mood Disorder
The three cardinal features of autism are 1) failure to develop communicative language, 2) failure to develop interpersonal interactions, and 3) failure to develop adaptive skills, the child being limited to a narrow range of repetitive and stereotyped actions and interests. How might these be related to primary mood disorder?
1) Language: A tentative analysis of the language deficit seen in young (2 to 7 years) autistic children follows, based on clinical observations. Clearly there is a major element of aphasia. Auditory processing is deficient leading to receptive aphasia. There is also an element of expressive aphasia, probably mixing inseparably with speech dyspraxia (thus articulatory difficulties), but also with problems in language syntax, semantics, and pragmatics. There is also, undoubtedly, an intellectual deficit that constrains language. But there are still other—mood and attentional—elements of the language deficit: mutism, negativism, and anxiety. I am referring to the child who vigilantly stares away from the examiner; who appears not to respond to voice or any stimuli, and does not speak—although at surprising and unexpected times may utter a clear sentence. After treatment with an SSRI (discussed below), or simply over time, mutism may diminish; speech utterance become freer; and syntax, semantics and pragmatics improve.
2 It should be evident that affect can overwhelm cognition. This is true in major mood disorder in adults; when severe enough to swamp the perception of reality, it constitutes psychosis. It should not be surprising that the same phenomenon can occur in much stronger form in the young, in whom cognition is only beginning to take root, while emotion and mood are vigorously active. And it may not be amiss to think of autism, in some cases, as a chronic psychosis.
2) Social interaction: Likewise, failure of social interaction has cognitive and mood aspects. In the cognitive domain, the autistic child doesn't learn social cues and rules (personal space, turn-taking, appropriateness, etc.). In the affective domain, negativism, stubbornness, opposition-defiance, aloofness, aggressiveness, and anxiety (social phobia) are features. Again, these can be modified pharmacologically in some cases.
3) Repetitive, stereotyped behavior; failure of adaptive behavior: The best example of concurrence between autistic features and familial mood disorder is afforded by recent studies of “insistence on sameness” in autistic children. Hollander and colleagues show a correlation between “insistence on sameness” in autistic children and a family history of obsessive-compulsive disorder.
14 And Shao and colleagues found a correlation between “insistence on sameness” and evidence of linkage to the GABRB3 gene in the chromosome 15q11–13 region.
15 These two important studies illustrate in a limited sphere our basic hypothesis: there is a correlation between familial mood disorder and features of autistic disorder, susceptible to genetic demonstration.
Other Clinical Features of Idiopathic Autism
The cardinal deficits of autism can be described in other terms, and additional features can be recognized, particularly in the “mood” subgroup. Factor analyses have defined independent subgroups of symptoms. One analysis identified social communication, emotional reactivity, social orienting, cognitive and behavioral consistency, and odd sensory exploration.
16 A separate factor analysis of social communication handicaps in autistic spectrum disorder identified three factors: joint attention, mood reciprocity, and theory of mind.
17 Both these analyses emphasize emotional and mood disturbances, which the traditional three-part description omits. Intense narrow fixated interests are characteristic of this form of autism. The low functioning child may be fascinated with hinges or pouring sand, the high functioning child with weather, arachnids, etc. This obsessive fixation is presumably linked to the need for sameness and difficulty with transitions these children commonly show, and is also related to obsessive-compulsive disorder. Likewise, motor stereotypies may be equated to compulsive actions, generally much accentuated by anxiety.
Social phobia is a major element in autism, as mentioned above. Similarly, sensitivity to sensory stimuli, such as sounds (e.g., vacuum cleaner, sirens), texture (e.g., velvet, glass), and touch (e.g., clothing tags, wrinkle in socks, texture of foods) may be phobic.
Mood Disorder in Idiopathic Autism
Heretofore we have attempted to show that a wide range of autistic symptoms may be understood as having fundamental similarities to major mood disorder. But we can look at the issue more directly. Many idiopathic autistic children betray frank symptoms of depression. They are anhedonic or frankly unhappy and lack a typical child's usual “joie de vivre.”
18 These children tend to withdraw from social contact and demonstrate frustration, negativism, and irritability. They display anxiety and phobias as noted above. Even the regression that commonly heralds the onset of autism may be interpreted as the onset of depression, characterized not only by loss of language, but by social withdrawal, loss of eye contact, moodiness, tantrums, fearfulness, and occasionally self-injurious behavior. Later, in some children, the full expression of manic depression may become evident and is marked by extreme cyclicity of moods, oppositional/defiant behavior, hyperexcitement (e.g., hyperactivity, aggressiveness, rage), and vegetative signs of mania (e.g., decreased sleep, excessive fluid intake), alternating with episodes of withdrawn depression. We have designated this latter group as childhood bipolar autistic disorder. In these children, the conflation of autism and manic depression (bipolar disorder) seems clear.
13The special case of Asperger's syndrome deserves attention. In our experience, as well as others, a majority of patients with Asperger's syndrome have clinically-evident major mood disorder by adolescence.
19 An unexpectedly high proportion of individuals from family pedigrees of Asperger's patients have a history of social phobia, social withdrawal, or hermitism.
Interconversion of Autism and Bipolar Disorder
Children with childhood bipolar autistic disorder, as mentioned above, are initially diagnosed as autistic and subsequently develop cycles of mood disorder typical of bipolar disorder. This occurs within a family setting of major mood disorder, usually including bipolar disorder in family members. Some of these children are eventually diagnosed as having Asperger's syndrome as they emerge from frank autism (as noted above, a large proportion of children and adolescents with Asperger's syndrome also have major mood disorder). Some emerge from autism entirely and can be diagnosed straightforwardly as bipolar. Special abilities, including the most salient savant abilities, seem to be closely related to this bipolar-autism nexus. Rare but informative autistic individuals are verbally and cognitively precocious before regressing and later become severely autistic, with bipolar disorder. Some hyperlexic children with autistic spectrum disorder develop Asperger's syndrome with bipolar disorder later in life. Some savant children lose their special abilities, becoming simply regressed autistics, often with bipolar disorder. Implicit in these case scenarios is the idea that bipolar disorder is often associated with cognitive hyperfunction—at times amounting to savant skills—which may later devolve into loss of function. As yet no adequate body of data exists to validate the foregoing.
II. Family History
Family history data repeatedly show that bipolar disorder and major depression increased in the families of autistic individuals.
2,5–8,13 Obsessive-compulsive disorder (OCD) and anxiety disorder are similarly said to be overrepresented in such families. Such family data are usually acquired by the family history method, which has high specificity and low sensitivity, with greater accuracy for major mood disorders, and less sensitivity in revealing less salient disorders. Familial studies of depressed probands vary in the absolute rates of mood disorders in relatives. A study of 215 mild and severely depressed nonbipolar major depressives and normal probands and 1,331 adult first-degree relatives found the rates of depression in relatives of the depressed subjects twofold to fivefold higher.
20 Similar increases in rates of major mood disorder have been found in the families of autistic children as compared with controls.
7 We have obtained family history data regarding the incidence of major mood disorder in families of autistic children in four separate series:
1) The first was carried out in Massachusetts between 1982 and 1986. The incidence of major mood disorders in the families of autistic individuals was significantly higher than published population rates of major mood disorders.
22) In North Carolina, between 1988 and 1990, we studied 40 autistic individuals (20 attributable to known neurological disease and 20 idiopathic). Family histories, using the family history method, without knowledge of the neurological status, showed a low incidence of major mood disorder in the neurological patients (only two had family members with major depression, none with bipolar disorder). In the idiopathic autistic patients, by contrast, major depression was found in 14 and bipolar disorder in 8 of twenty families.
23) In subsequent years, we studied a large series of autistic children seen clinically. Family history was ascertained during successive clinical visits. This resulted in a much more complete and accurate picture of family psychopathology than could be obtained in a single visit, or by an investigator who was not the responsible physician. Seventy percent of families of idiopathic autistic children had one or more family member with major mood disorder (included were first and second degree relatives of the parents).
44) Between 1995 and 2002, we acquired another series of patients included in our study of fluoxetine treatment for young autistic spectrum children. We determined family history data as before and sought information about family members with special intellectual abilities or attainments, inspired by observing such individuals in many of the families. The abilities most often were scientific, mathematical, or computational but included others (e.g., professor of philosophy, professional musician). Analysis revealed a strong correlation among three groups: autistic probands responding to fluoxetine, family members with major mood disorder (especially bipolar disorder), and family members with special intellectual abilities. In this study, history of major mood disorder (in first- and second-degree parental relatives) was assessed in 151 families. One hundred and eleven families (74%) had a history of major depression (in 102) and/or bipolar disorder (in 52).
21III. Pharmacologic Response
There is increasing recognition that childhood autism can be favorably modified by appropriate medication. Medications that proved to be beneficial are the same as those used for mood disorder in the nonautistic population, namely SSRIs (selective serotonin reuptake inhibitors), atypical antipsychotics, and mood stabilizers (lithium or antiepileptic agents). This does not prove that autism and mood disorder are the same, but strongly suggests that their neurotransmitter and receptor characteristics must be similar. Authors commonly state that neuropharmacologic agents in autism don't treat core symptoms, but only modify comorbid or incidental features. However, our clinical experience suggests that the frequent improvement in socialization, language, adaptive skills, and mood indicates that effective medication does modify, though not cure, the core symptoms of autism.
We have had extensive experience treating young idiopathic autistic children with fluoxetine over the past 8 years.
21 This experience has been limited to open trials, continued for long durations (see below), controlled by intermittent drug holidays and compared to trials of other medications in the same children. That experience is summarized here. Of 129 children in whom treatment could be evaluated: 22 (17%) had an excellent response; 67 (52%) had a good response; 10 (8%) had a fair response, and 30 (23%) had a poor response. The mean age starting treatment was 54 months (4½ years). The mean length of treatment to date is 36.6 months (range 13 to 76 months) for excellent responders, and 32.3 months (range 5 to 76 months) for good responders.
Thirty children were maintained on a regimen of fluoxetine for 36 months or more (range 36 to 76 months). Twenty were good and 10 were excellent responders. Twenty-five were continuing treatment when last encountered. Twenty had trials of discontinuing treatment, of whom 18 resumed treatment after clinical regression; one had no regression; one was lost to follow-up. Six families refused discontinuation trials, and data was unavailable in four. With regard to educational placement, 14 were in regular class (some with an aide), five in autistic class, three in other special class, and five unknown.
Five boys were later diagnosed independently with bipolar disorder; four of these were no longer considered autistic, and were in regular (N=3) or learning disability (N=1) classes; one was still clearly autistic and in an autistic class. Four were treated with lithium, one with valproate.
Response to fluoxetine correlated with major mood disorder in the proband's family: of probands with a positive family history, 67 of 86 had a good/excellent response (78%), while of those with a negative family history, 12 of 31 (39%) had a good/excellent response. (χ2 = 17.2, p<0.001) Fluoxetine response likewise correlated with special intellectual abilities in the proband's family: 52 of 68 probands with a positive family history of special abilities (76%) had good/excellent fluoxetine response, while 32 of 53 with no family history of special abilities (60%) had a good/excellent response. (χ2 = 3.8, p = 0.05) However, selecting only those with excellent fluoxetine responses, 21 of 22 had family history of special ability.
In light of the above, it is not surprising that a strong correlation was found between major mood disorder and special intellectual ability in families. (χ2 = 20.8, p<0.0001) If restricted to bipolar disorder, the correlation with special intellectual ability is even stronger. (χ2 = 38, p<0.00001) In addition, hyperlexia in the probands (defined as precocious interest in letters and numbers) correlated robustly with treatment response. Of children with autistic spectrum disorder and hyperlexia, 24 of 25 showed an excellent (N = 10) or good (N = 14) response.
Risperidone, in a short-term (8 weeks) controlled study, improved serious behavioral problems of autism, particularly severe tantrums, aggression, or self-injurious behavior.
22 Anecdotal observations indicate similar improvement with other atypical antipsychotic agents; we have been impressed with parental reports of striking cognitive improvement in a few cases with aripiprazole. We have found lithium beneficial for individuals with childhood bipolar autistic disorder.
13 Divalproex sodium has been useful, even in the absence of seizures or EEG abnormalities, suggesting it was acting as a mood stabilizer.
14 McDougle and Posey have suggested that the combination of a D2 antagonist, 5HT1A agonist, and 5HT2A antagonist may give optimal benefit in autism.
23In summary, the agents shown beneficial for autism are exactly the same as those used in treatment of bipolar and related mood disorders, strengthening the concept that the neurotransmitter/receptor abnormalities are similar in both entities.
IV. Neuroimaging Studies
The current neuroimaging revolution promises to yield great insight into cognitive and mood disorders. We may ask whether available data give any insight regarding similarities or differences between autism and mood disorders.
Neuroimaging in Autism
For a review of functional neuroimaging in autism see Boddeart and Zilbovicius.
24 Perhaps the single most important finding in the study of autism has been the PET study of Chugani and colleagues
25 using 11C-alpha-methyltryptophan, a serotonin precursor used to measure local serotonin synthesis in the brain. In seven autistic boys (5 to 7 years of age), the authors found markedly decreased serotonin synthesis in the left hemisphere (most marked in frontal cortex, but also decreased in temporal and parietal cortex and in subcortical structures), along with increased serotonin synthesis in the right (contralateral) cerebellum, in five; in two (whose clinical picture reportedly did not differ) the asymmetry was reversed. Further studies, including girls, indicated that young autistic children showed a delay in the development of serotonin synthesis in frontal cortex compared to normal children; this was often bilateral, especially in girls.
26 Because serotonin has an important role in cortical development, especially in controlling branching and growth of corticopetal axons, abnormalities of serotonin might be implicated in the excessive growth of brain in autistic children between birth and two years;
27 however this is only speculative. In autism, SPECT scanning revealed decreased rCBF in left prefrontal, medial prefrontal, and anterior cingulate gyrus, and in left amygdala; also decreased rCBF in thalamo-cortical systems.
36 Adults with autistic disorder, studied with functional MRI, did not activate a cortical ‘face area’ when explicitly appraising expressions, nor the left amygdala region and left cerebellum when implicitly processing emotional facial expressions.
37 Baron-Cohen and colleagues,
38 using functional MRI, studied social intelligence (so-called “theory of mind”) in adult autistic men, finding that they activated the left prefrontal cortex less than normals, and did not activate the left amygdala.
These studies raise three points pertinent here:
1) The role of serotonin in autism and depression; 2) the lateralization of serotonin synthesis in autism, which appears to parallel the abnormal lateralization of function, and evidence of similar lateralization in depression; and 3) the role of cerebello-cortical systems.
Neuroimaging in Depression:
Do the neuroimaging data in depression correspond with those for autism with respect to the key issues of serotonin abnormalities, hemispheric lateralization, and cerebellar involvement? PET studies using receptor ligands suggest that major depressive disorder is associated with decreased serotonin neurotransmission. Decreased 5HT1A receptors were found in limbic and neocortical areas, specifically in mesiotemporal, occipital and parietal areas.
29,30 These abnormalities were most prominent in bipolar depressives and unipolar depressives who had bipolar relatives. Decreased 5HT2 receptors were also found in depression.
31 In these studies, no clear lateralization was observed.
There is substantial evidence of abnormal blood flow and metabolism in depression, with a characteristic pattern affecting prefrontal cortex, anterior cingulate cortex, and mesolimbic areas (amygdala and hippocampus). Left-sided predominance of abnormalities has been described for prefrontal cortex
32,33 and amygdala.
32,34 Another study of cerebral metabolism in major depression, using 18FDG PET, revealed low left hippocampal metabolism in subjects with MDD, or with concurrent OCD and MDD. Hippocampal metabolism was negatively correlated with depression severity across all subjects.
35Comparison of Neuroimaging in Depression and Autism
A review of neuroimaging studies in neuropsychiatric disorders including autism and depression (plus schizophrenia and post-traumatic stress disorder), emphasizing social cognition, concluded that all are associated with dysfunction in the amygdala and dorsal cingulate gyrus.
39 Proton magnetic resonance (MR) spectroscopy in bipolar disorder revealed decreased N-acetylaspartate and choline concentrations in frontal lobe.
40 In autistic children, concentration of N-acetylaspartate and the signal intensity were reduced significantly in the left amygdaloid-hippocampal region and the left cerebellar hemisphere.
28 Interestingly, in a separate study subjects with Asperger's syndrome were found to have higher prefrontal concentrations of NAA, choline, and creatine; the higher NAA correlated with obsessive behavior.
41 It is difficult to compare findings in autism and depression, because of differences in techniques, age, and other factors. However, the findings taken together support the following interpretations:
1) Both autism and depression appear to be characterized by disturbances in serotonin metabolism: in autism by decreased serotonin synthesis and a disturbed trajectory of developmental change of serotonin synthesis; in depression, by decreased 5HT1A and 5HT2A receptors. The exact relationship between these two observations is uncertain.
2) Both autism and depression primarily involve the left hemisphere. (The abnormal lateralization of function seen in autism [as shown, for example, by the verbal/performance split in IQ testing] tends to fade after age 5 and is not prominent in girls, in whom bilateral decreases in serotonin synthesis are generally seen. Thus, comparisons above age 5 and in girls may mute the question of laterality.)
3) Autism and depression show abnormal function in congruent areas in the brain, principally prefrontal cortex (left more than right), left amygdala and hippocampus, cingulate cortex, and cerebellum. Both may be said to involve the “social brain.”
Notably, a pilot study indicated that multi-episode bipolar patients have smaller V3 segment of the cerebellar vermis (lobules VIII–X) than first-episode bipolar patients or healthy volunteers.
42 Reduction in the size of cerebellar vermis has been widely reported in autism, not without controversy;
43 here is a suggestion that a similar abnormality may be seen in bipolar disorder, occurring later and more mildly.
V. Neuropsychological Observations
The cognitive deficit in autism is prominent and well recognized. Are there similar cognitive disturbances in major mood disorder?
Disordered Cognition in Bipolar Disorder
A recent review of cognitive impairment in bipolar disorder concluded that “changes in the fluency of thought and speech, learning and memory impairment, and disturbances in associational patterns and attentional processes are as fundamental to depression and mania as are changes in mood and behavior. Moreover, a significant number of bipolar patients show persistent cognitive deficits during remission from mood symptoms. Most studies point at the presence of diffuse cognitive dysfunction during the acute phases of bipolar illness. Most of the deficits seem to remit during periods of euthymia, but some may persist in approximately one third of bipolar patients.”
44 Verbal memory and sustained attention were abnormal in symptomatic bipolar patients, and persisted in remission. All aspects of executive function (planning, abstract concept formation, set shifting) were impaired in symptomatic patients but could be normal or still impaired in fully recovered patients.
45 The parallels with autistic individuals with regard to attention deficits, impairment of executive functions, and verbal memory disturbance—as well as diffuse cognitive impairment—are evident.
Cognitive Competence in Families of Autistic and Bipolar Probands
Closely related to the cognitive disorders in autistic and bipolar individuals is the question of cognitive competence in other family members. The evidence seems clear that special abilities occur with increased incidence in families of autistic and of bipolar probands, and that the special abilities are similar for each entity. This is a most intriguing observation: it may have something to teach us about the genetics and evolution of human intelligence. It suggests that autism and bipolar disorder represent the reverse genetic side of the special abilities of other family members.
Special Ability in Families of Bipolar Probands
An association of bipolar disorder with high intelligence and social achievement among family members has been documented.
46 The socioeconomic advantage previously associated with mood disorder in general may be limited to the bipolar forms, and to family members, not the affected probands. A study of the occupational levels of unipolar and bipolar probands and their relatives revealed a social advantage both for male and female members of bipolar families. An overrepresentation was found in the higher occupational class in bipolar probands' brothers and children.
47 Offspring of bipolar patients have been rated on their personal and social resources. Positive resource profiles were related to psychiatric well-being in the offspring. Nondisordered offspring demonstrated a strikingly positive profile of personal resources as well as a wide range of peer, sibling, and other kin supporters.
48Special Abilities in Families of Autistic Probands
In 154 families of our autistic probands, we found that 52% of the families had one or more individuals having special intellectual abilities.
21 These abilities often involve computational skills; more broadly, they reflect a capacity to organize large amounts of data. These abilities are best illustrated by the occupations represented among individuals possessing such abilities: statistician, mathematician, linguist, physicist, engineer, physician, research radiologist, metallurgical scientist, biochemist, financial analyst, computer architect, actuary, professional violinist, psychologist, nuclear power plant operator, director of research and development, biotech executive; professor of philosophy, political science, physics, biology, etc.
We demonstrated above that this kind of intellectual prowess correlates strongly with a family history of major mood disorder (especially bipolar disorder) and also with fluoxetine response of the autistic proband.
VI. Hemispheric Lateralization of Function
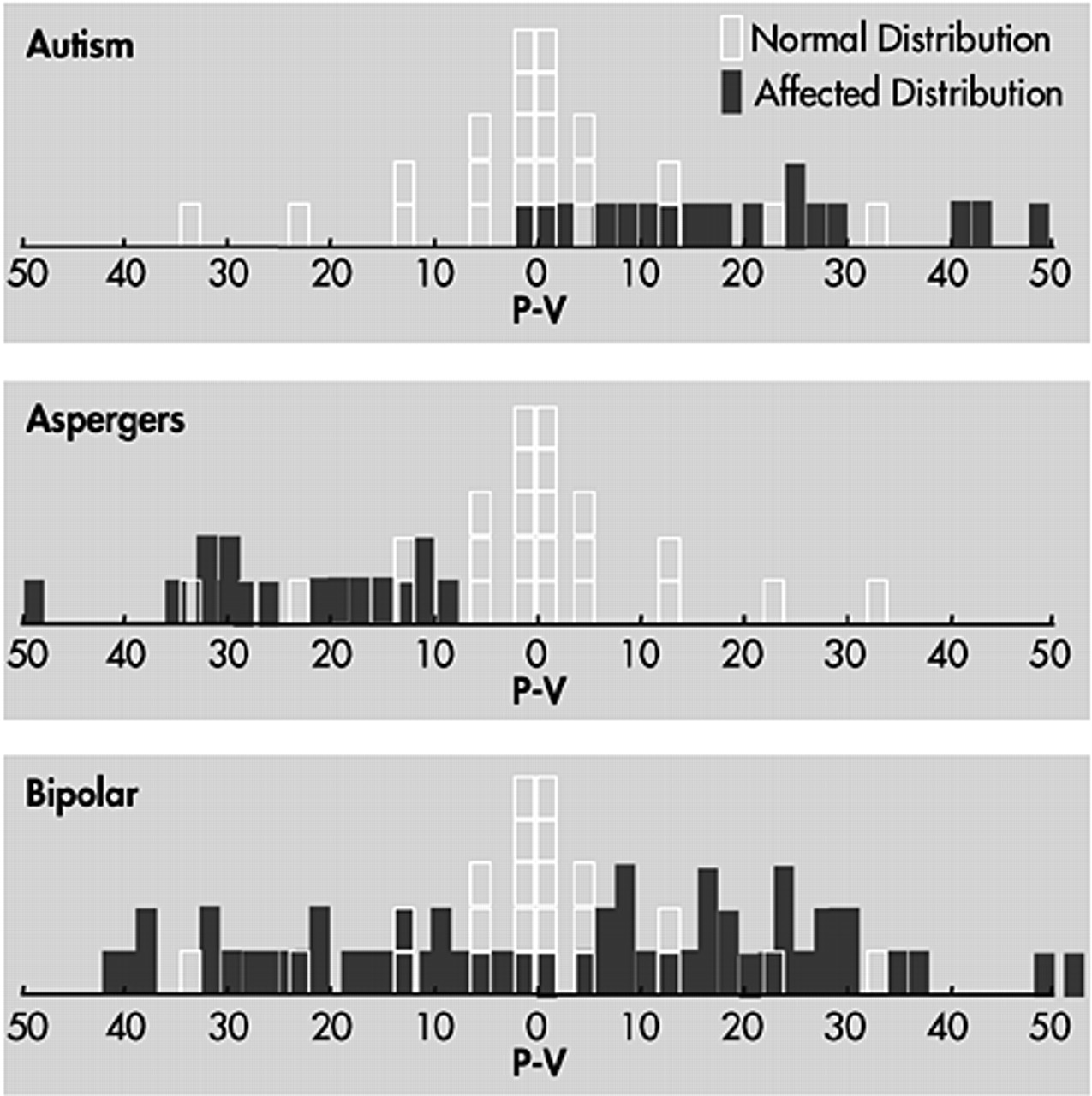
Both autism and bipolar disorder evince interesting aberrations of hemispheric lateralization, which may prove to be fundamental to the pathogenesis of these disorders. It is beyond the scope of this review to survey the question of hemispheric lateralization in detail. We may summarize our observations with a chart (
Figure 1. Children with bipolar disorder typically show excessive spread between their verbal and performance scores on WISC-R (or WISC-III) testing. Virtually none of the bipolar children show equal verbal and performance scores; most are strongly skewed to one side or the other. Shown above the bipolar children in the Figure are the performance minus verbal scores measured in a group of our patients with autism or Asperger's syndrome. Not surprisingly, the autistic children show a pattern of higher performance than verbal scores, and the Asperger's children show the opposite.
Lateralization of function has been compared in individuals with high-functioning autism and with Asperger's disorder. The autism group displayed deficiencies in right hemispace (and by implication, left hemisphere) performance on executive function tasks; however, they demonstrated normal lateralization effects on nonexecutive, visual-perceptual tasks. In contrast, the Asperger's group showed normal laterality effects compared to controls on both executive and nonexecutive function tasks.
49Hemispheric Lateralization in Bipolar Disorder
These results in children with bipolar disorder show skews in hemispheric function that can favor either the left or right hemisphere. Some have hypothesized that anomalous cerebral asymmetry is a feature of psychotic disorders generally.
50 Most studies of adult bipolar disorder relate manic excitement to right hemisphere dysfunction (hyperactivation) and depression to dysfunction of the left hemisphere.
51 Bipolar mood disorder, closely resembling functional bipolar disorder, may occur in patients with cerebrovascular lesions. In most such patients, this secondary bipolar disorder is associated with right hemisphere lesions that involve subcortical and midline structures,
52 possibly releasing right cortical hyperactivity.
Neurocognitive performance has been found significantly impaired in euthymic adult bipolar patients in a range of visuospatial tasks.
53 The abnormalities were not related to age of onset, duration of illness, or medication; but they were associated with the number of previous affective episodes. In contrast, in childhood bipolar disorder we often find superior visuospatial function, attested by the interest and skill these children often show in drawing, as well as by formal neuropsychological testing (see above). This apparent paradox of excellent visuospatial function in the young bipolar child, and impaired visuospatial function in bipolar adults, may suggest that this cognitive function deteriorates over time as a result of repeated mood episodes. (An analogous case in the verbal realm might be the rare young autistic child who develops precocious verbal abilities, which later disappear when the child regresses.) While there is no adequate body of data on these questions, they introduce the concept that cerebral hyperfunction is a part of bipolar disorder, and is vulnerable to exhaustion or self-destruction. Again, the parallels between autism and bipolar mood disorder seem natural and ineluctable: hyperfunction, skewed hemispheric lateralization of function, and loss of function are common to both entities. (In this regard, recall the finding that the posterior cerebellum has been found to show atrophy in bipolar disorder in proportion to the number of mood episodes;
42 and that in autism the posterior cerebellum shows early atrophy that progresses over years.)
43Special Abilities in Autistic and Bipolar Probands
The autistic savant must be mentioned in connection with abnormal hemispheric lateralization of function and special cognitive abilities. Savant characteristics occur primarily in autistic spectrum disorders, but they also occur in individuals with bipolar disorder who are not autistic—unless one takes the special or precocious abilities of some bipolar individuals as proof of Asperger's syndrome, which is tautological. Savant abilities are generally considered right-hemisphere mediated abilities. We categorized savant characteristics occurring in a group of bipolar children.
3 It is not rare to see bipolar individuals with special talent in drawing, photographic memory, mathematical or musical talent, poetic talent, unusual memory or other similar ability. These same abilities occur, of course, in autistic individuals. It is our belief, from extensive clinical experience, that these savant features are similar in kind in bipolar disorder and in autism, and that the two entities may interconvert.
Hemispheric Lateralization of EEG Activation in Autistic Infants and Infants of Depressed Mothers
Autistic children exhibit reduced EEG power in the frontal and temporal regions, but not in the parietal region, more prominent in the left hemisphere.
54 Depressed adults also exhibit reduced left frontal electroencephalographic activity. Transcranial magnetic stimulation in patients with major depressive disorder showed significant interhemispheric differences in motor cortical excitability, with lower excitability on the left hemisphere; such differences were absent in controls.
55 Dawson and colleagues found that 13-to 15-month-old infants of depressed mothers exhibited reduced left frontal lobe EEG activity compared to infants of nondepressed mothers. The reductions were greater in infants of mothers with major depressive disorder.
56VII. Neuropathology
Neuropathological changes described in autism include macroencephaly, acceleration and then deceleration in brain growth, increased neuronal packing and decreased cell size in the limbic system, decreased Purkinje cell number in the cerebellum, and abnormalities in organization of the cortical minicolumn.
57 Additionally, a new neuropathology of psychosis is emerging, focused on synapses, synaptogenesis, and synaptic plasticity,
58 which has revealed a convergence in the pathological findings in schizophrenia, bipolar disorder, and autism.
Abnormal markers in prefrontal cortical areas in postmortem specimens from patients with schizophrenia and bipolar disorder have been summarized.
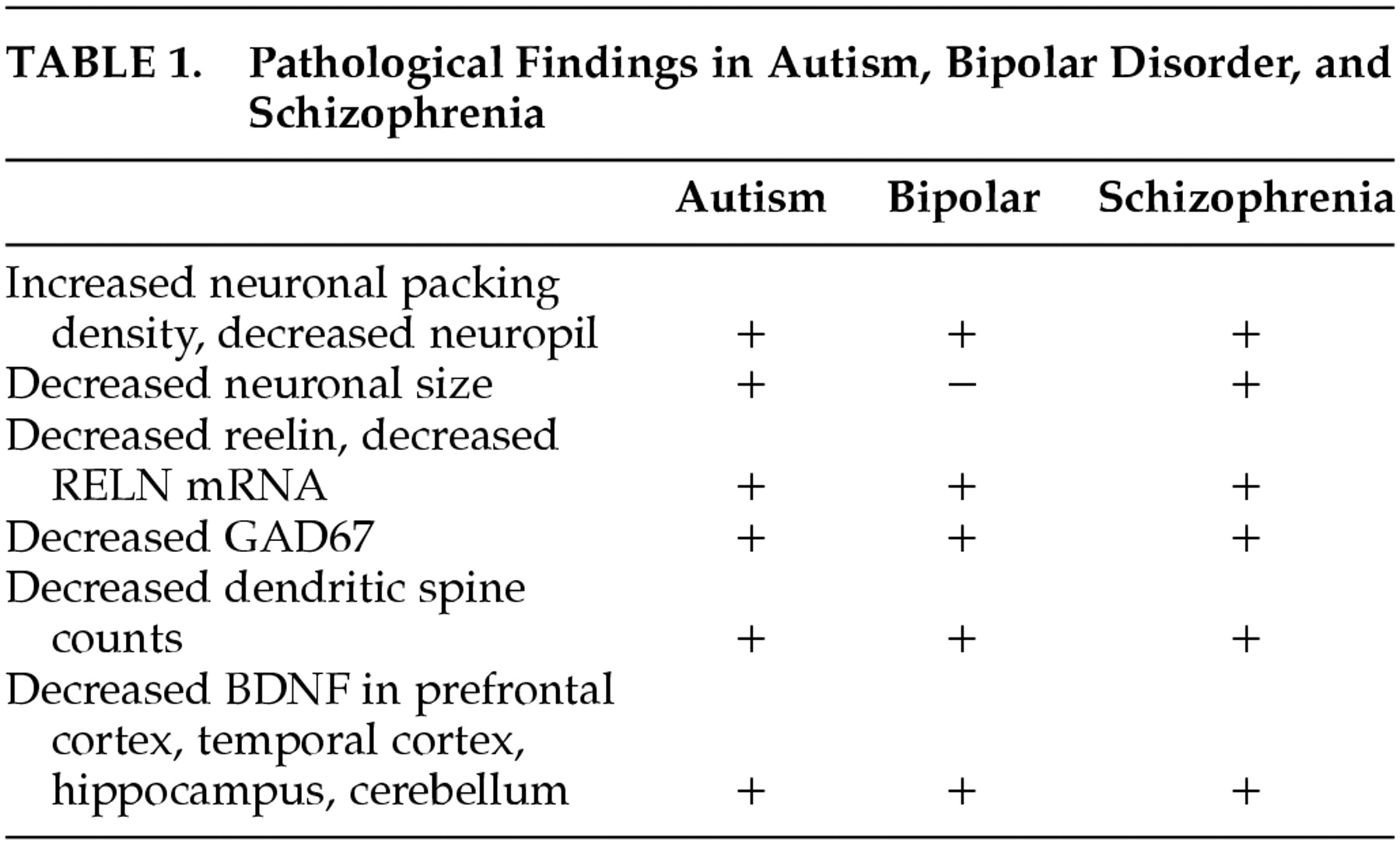
59 The markers pertained to a variety of neural systems and processes including neuronal plasticity, neurotransmission, signal transduction, inhibitory interneuron function and glial cells. The most salient markers pertained to aspects of glutamatergic, GABAergic, and dopaminergic neurotransmission. The neuropathology of schizophrenia includes decreased reelin and glutamic acid decarboxylase (GAD67) expression, increased neuronal packing density and decreased neuropil and dendritic spine number.
60Decreased dendritic spine counts (40%) are found in frontal cortex in brains of patients with bipolar disorder, schizophrenia,
61 and autism.
62 Decreased BDNF and decreased reelin (both involved in synaptic and dendritic spine plasticity) have been described in prefrontal cortex in autism, bipolar disorder, and schizophrenia.
63Reelin has been postulated to be centrally important in schizophrenia as well as bipolar disorder and autism. Postmortem studies of prefrontal cortices, temporal cortices, hippocampi, caudate, and cerebella of schizophrenia patients and their matched nonpsychiatric subjects were compared for reelin mRNA and reelin protein content. In all of the brain areas studied, reelin and its mRNA were significantly decreased (approximately 50%) in patients with schizophrenia.
64 These findings parallel those found in autism.
65 The
Table summarizes reported similarities in pathological findings in autism, bipolar disorder and schizophrenia.
VIII. Neurochemical Studies
Gamma-aminobutyric acid (GABA) receptor abnormalities may determine mood disorders. One putative genetic site determining bipolar disorder is Xq28, a site for GABRA3, a GABA receptor subunit; another possible example is GABRB3 on chromosome 15q11–13 (see below). The GABergic and serotonergic systems have extensive and complex interactions, which may be important both in autism and in bipolar disorder.
67,68 These interactions could account for the efficacy of SSRIs in treatment of mood disorder and autism.
69,21Reelin and GAD67 are colocalized in GABergic interneurons. Reelin is found at GABergic synapses, co-localized with clusters of alpha3integrin molecules in post-synaptic densities located on apical dendritic spines. Reelin and integrin activate a cascade that modulates cytoskeletal elements thought to mediate synaptic and spine plasticity.
60 Closely related to this sequence is the action of antidepressants on TrkB neurotrophin receptor: activation of the TrkB neurotrophin receptor is required for antidepressant-induced behavioral effects, in a BDNF-dependent manner.
70 Reelin is an activator of BDNF. These interactions are presumably important in SSRI-responsive depression and autism.
Mutations of X-linked genes encoding neuroligins NLGN3 and NLGN4 have been associated with autism; these mutations affect cell-adhesion molecules localized at synapses, and suggest that various defects of synaptogenesis may predispose to autism.
71 The foregoing is only a sketchy look at a rapidly advancing area of investigation. It suffices, however, to suggest a congruence of ideas about the pathogenesis of schizophrenia, bipolar disorder, and autism.
IX. Genetics
No exhaustive review of genetic studies in autism is contemplated here. We must bear in mind that autism is surely genetically heterogeneous. We have focused on that group of autistic children characterized by responsiveness to SSRIs with a family history of major mood disorder and often special intellectual abilities. Such children commonly have a history of regression, are higher-functioning (in some cases demonstrating hyperlexia and/or other special intellectual characteristics) and have prominent obsessive, mood, and dyspractic features. We estimate this group to comprise about two-thirds of idiopathic autistic individuals (and suggest that the other third may be characterized by prominent language disability, lower function overall, a family history including individuals demonstrating the “broader autistic phenotype”, no association with familial mood disorder, and poor response to SSRIs.)
7,8 This grouping is tentative, but seems useful. We will consider genetic studies only of the first group; we suggest that the second group may be associated with a different set of genes more directly related to speech and language (e.g. 7q, 2q, 13q).
An important subgroup of autistic spectrum disorder is linked to chromosome 15q11–13.
72 The most frequent chromosomal abnormality seen in autistic populations involves duplication of sequences in a region on the proximal part of the long arm of chromosome 15, specifically the interval 15q11–13. These duplications are associated with substantial risk of autism when derived from maternal but not paternal chromosomes. This is the interval involved in Prader-Willi syndrome and Angelman syndrome. Prader-Willi and Angelman's patients often show phenotypic overlap with autism and/or have severe mood disorder (and in Prader-Willi syndrome show a characteristic response to fluoxetine, which they tolerate in high doses [unpublished]). Oculocutaneous albinism (OCA2) in this region has co-occurred with autism in several published cases
73 and three of ours [unpublished].
Results of genomic linkage studies with regard to chromosome 15q11–13 in autism are mixed.
74 Existing genetic data converge on a cluster of three GABAA receptor subunit genes (beta3, alpha5, and gamma3), most specifically an association with a marker in the GABRB3 (beta3) gene.
75 Linkage data selecting for a specific phenotypic subtype trait (“insistence on sameness”) to enhance homogeneity showed increased linkage evidence for the 15q11–13 region, at the GABRB3 locus.
15 The authors suggest that “insistence on sameness” is a trait related to OCD, and thus to mood disorder (OCD has a large comorbidity with bipolar disorder
76 ). A similar strategy based on savant skills in autism families improved evidence of genetic linkage to 15q11–13
77 (recall that we have related savant skills to bipolar disorder).
3,13A convergence of Prader-Willi syndrome, autism, and affective disorder demands special notice. Prader-Willi, caused by failure of expression of paternally-derived genes in the 15q11-13 region, is associated with severe behavioral problems (apart from hyperphagia) including compulsive behaviors including hoarding leading to full-blown obsessive-compulsive disorder; tantrums, oppositionality, and aggression; and in young adults with uniparental disomy, severe affective disorder with psychotic features.
78,79Also, persons with Prader-Willi syndrome show strikingly superior ability, compared to normal controls, in jigsaw puzzle proficiency,
80 and Prader-Willi individuals with maternal uniparental disomy demonstrated superior visual recognition memory compared to normals.
81 This suggests a gain-of-function mutation in Prader-Willi that is reminiscent of the superior visual-spatial skills seen in some autistic children. As a final point, Prader-Willi syndrome may show grandmatrilineal inheritance,
82,83meaning that a genetic defect deriving from the grandmother will not be expressed in her sons, but in the sons’ children (because the imprinted maternally-derived 15q11-13 genes are not expressed in her son, but will be transmitted unimprinted by the father, to be expressed in his children with 50% probability). Lest this seem too far removed from our subject, let me describe a family in which two young cousins have classical autism: their great grandmother has superior intelligence and a history of hoarding and rigidity. Her daughter is an artist and hoarder. The latter has two sons; one is an artist who had precocious talent at age 5 years, has photographic memory, hoarding behavior, and bipolar disorder requiring hospitalization and treatment with lithium. He has a 7-year-old boy with classic autism and oculocutaneous albinism (see above). The second son has special intellectual ability (computer designer), may be considered as having Asperger’s syndrome, and has a son with autism very similar to that of his cousin. This vignette illustrates the concurrence of obsessive-compulsive behavior, bipolar disorder, special intellectual ability including eidetic memory, oculocutaneous albinism, and autism, with grandmatrilineal inheritance. Formal linkage studies in this family support 15q11-13 linkage. This clinical pedigree virtually speaks for itself. It suggests that clarification of genetic mechanisms, especially imprinting, in the Prader-Willi/Angelman region may have direct pertinence for autism as well as mood disorder. The presence of special visual-spatial skills in Prader-Willi syndrome and the finding, mentioned above, that subsetting of autistic families based on savant skills improved evidence of genetic linkage to 15q11-13, strengthen the concept that a gain-of-function mutation in this region of the genome contributes to autism, Prader-Willi syndrome, and bipolar disorder.
Genetic linkage of bipolar disorder has been complex and vexing, and cannot be reviewed here. It seems justifiable, in the present context, to mention results suggesting linkage of bipolar disorder to chromosome 15q11–13, without mentioning all other putative linkages. Papadimitriou and colleagues reported an association between the GABA(A) receptor alpha5 subunit gene locus (GABRA5) and bipolar mood disorder.
84 They found no association between bipolar mood illness and GABRB3, and suggested GABRA5 may be the gene actually associated with the disorder.
85 Earlier studies were inconclusive; two did not find a linkage of bipolar disorder to chromosome 15,
86,87 while another did.
88 A single case study described autism and bipolar disorder in a woman with chromosome 15 deletion.
89At the chromosome 15q11–13 linkage site, likely candidate genes include GABA(A) receptor subunits (see above). Given the strong interaction of the serotonergic system on the GABergic system (discussed above), a therapeutic effect of serotonergic agents (such as SSRIs) on a disorder primarily determined by a GABA(A) receptor subunit abnormality is logical, but speculative.
The Serotonin Transporter Gene in Depression and Autism
Intense interest in the serotonin transporter gene (5-HTT), particularly on the role of the
short and
long allele polymorphisms in the promoter region of the gene, has explicitly joined research on depression with that on autism. Association has been demonstrated between the 5-HTT promoter polymorphism variation and mood response during tryptophan depletion,
90 in amygdalar neuronal activity in response to fearful stimuli,
91 and in the influence of life stress on occurrence of depression.
92 Thus a role for 5-HTT promoter polymorphisms in depression and anxiety seems assured.
Equal interest has focused on a possible association of the 5-HTT promoter polymorphisms with autism. They appear not to be major determinants of the hyperserotonemia seen in autism.
93,94 Transmission disequilibrium in autistic disorder at this gene locus has been sought with uncertain results; the most recent work showed nominally significant evidence of transmission disequilibrium.
95 Thus while a role for this polymorphism seems clear in depression, it remains murky for autism. Notably, however, the effect of this polymorphism in depression reveals itself only in the presence of life stress or biological stress (tryptophan depletion); perhaps a similar stress is required to show a role for this genetic polymorphism in autism.
Conclusions
Among individuals with idiopathic autism, an important subgroup (corresponding to classical autism) occurs in families with a high incidence of major mood disorder in family members. We found that fluoxetine response of autistic probands correlated strongly with such a positive family history as well as with special intellectual abilities in family members.
These with other findings support a hypothesis that autism in these families is etiologically and genetically related to major mood disorder. This hypothesis suggests a biological and medical context for autism—or an important subgroup of autism—that has been missing. It further proposes that the biological concept of familial mood disorders be broadened to include cognitive and neurodevelopmental dimensions. In general, agents (e.g., genes, hormones, neurotrophic factors, neurotransmitters) active in brain during development may produce very different effects than the same agents acting in adulthood. Genes related to major mood disorder may produce a different phenotype—that of autism—if active in early life. It is equally likely that major mood disorder and the related subgroup of autism could share one or more genetic determinants but differ in others. I have tried to test this hypothesis by juxtaposing current knowledge of both disorders and examining similarities and differences between them in all aspects: clinical, familial, pharmacological, cognitive, neuroimaging, biochemical, pathological, and genetic. We find robust correspondence at many junctures between autism and major mood disorder and suggestively analogous correspondences at others. This juxtaposition and comparison fails to disprove the hypothesis. The definitive test will be genetic. We hope this discussion may help establish a basis for further genetic studies.
ACKNOWLEDGMENTS
The author thanks the John A. Jones Family Trust and the Charles and Sara Goldberg Trust for their support and Sherri Burch for assistance with manuscript preparation. The help and encouragement over many years from many patients and their families is gratefully acknowledged.