Despite a similar lifetime prevalence of major depressive disorder across racial and ethnic groups in the United States, patients from minority groups typically have poorer access to mental health care and are less likely than whites to receive either pharmacological or nonpharmacological treatment (
1–
7). Many, but not all, studies of depression treatment in naturalistic settings have shown poorer outcomes for patients of minority backgrounds compared with whites (
3,
4,
6,
8,
9). On the other hand, treatment outcomes are similar between minority and white groups in clinical trials using protocol-driven and measurement-based care, particularly after adjustment for baseline sociodemographic and clinical variables (
10–
15).
In the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, blacks (particularly young men) had the highest attrition rates and the least robust response to initial treatment with citalopram; however, most outcome differences were not significant after adjustment for baseline sociodemographic and clinical variables (
14,
16). Approximately 30% of participants achieved remission with citalopram; an additional 35% achieved remission after adding another antidepressant or buspirone, a finding suggesting that two antidepressants might be more effective than one (
13), which has been supported by recent reports (
17–
19). These studies raise the question of whether initiating treatment with two antidepressants would lead to a quicker or more robust outcome for patients with depression and whether outcomes would be affected by race or ethnicity.
The Combining Medications to Enhance Depression Outcomes (CO-MED;
www.co-med.org) study used a patient cohort different from the cohort for STAR*D to compare outcomes from use of two different antidepressant combinations with outcomes from use of a single antidepressant in a 12-week acute phase and subsequent continuation phase (
20). This article reports the findings from a secondary analysis of the data, which examined associations between race-ethnicity and outcome in a manner similar to that done in STAR*D.
Methods
Study overview
CO-MED was a seven-month single-blind, randomized trial that compared the efficacy of two antidepressant medication combinations with the selective serotonin reuptake inhibitor (SSRI) escitalopram plus a placebo (1:1:1 ratio). Outpatients with nonpsychotic major depressive disorder were recruited from six primary and nine psychiatric care sites. Sites were selected to ensure adequate patient flow and minority representationand to represent primary and psychiatric care. A sample size of 660 outpatients was chosen to allow detection of roughly a 15% difference in remission rate between each antidepressant combination and escitalopram plus placebo (with an expected remission rate of 35%). This difference was viewed as sufficiently large to affect practice, because it approximates the benefit of a single antidepressant over placebo in successful antidepressant registration trials.
Recruitment
Treatment-seeking patients were enrolled from March 2008 through September 2009. Potential participants were screened with each site's standard procedure (variable across sites), which could include using questions from the Patient Health Questionnaire (
21,
22) or physician and nurse queries during routine clinical visits. Patients meeting screening criteria were identified to their study clinicians and met with the site's clinical research coordinator, who explained the protocol and obtained written informed consent before proceeding.
Participants
Broad inclusion and minimal exclusion criteria were used to ensure a reasonably representative sample. Outpatient enrollees, 18–75 years of age, met
DSM-IV-TR (
23) criteria for either recurrent (one or more prior major depressive episodes) or chronic major depressive disorder (current major depressive episode for two or more years) based on a clinical interview and confirmed with a
DSM-IV major depressive disorder symptom checklist. Eligible participants had to be in the index episode for a minimum of two months to reduce the likelihood of placebo response and had to have a score ≥16 on the 17-Item Hamilton Rating Scale for Depression (HRSD-17) (
24). A list of exclusion criteria is available at
www.co-med.org.
The study protocol was developed according to the principles of the Declaration of Helsinki. The protocol and all consent and study procedures were approved by the institutional review boards at the National Coordinating Center (University of Texas Southwestern Medical Center), the University of Pittsburgh Data Coordinating Center, each participating regional center, and all clinical sites.
Baseline data
Sociodemographic and illness features were gathered at baseline. The anxiety subscales of the baseline HRSD-17 and the 30-item Inventory of Depressive Symptomatology-Clinician-Rated (IDS-C-30) (
25–
29) established the presence of anxious features. The self-report Psychiatric Diagnostic Screening Questionnaire (PDSQ) (
30,
31) established the presence of current axis I disorders with 90% specificity (
32). The Concise Health Risk Tracking-self-report scale (
33) established degree of suicidal ideation, the Altman Self-Rating Mania Scale (ASRMS) (
34) established the presence of manic symptoms, and the Cognitive and Physical Functioning Questionnaire (
35) measured functioning. The Self-Administered Comorbidity Questionnaire (SCQ) (
36) established the presence, severity, and functional impact of a range of general medical comorbidities.
Data on race and ethnicity were collected by self-report. Participants could select one or more of the following choices: Asian American, black, Native American, Pacific Islander, white, or other; in addition they could identify whether they were Hispanic.
Antidepressant treatment
We chose a 12-week study period (acute phase) so thatmaximal doses (if needed) could be delivered for at least four weeks, most participants whose depression could remit would do so without an excessively long trial, and attrition might be minimized. Treatment visits were planned at baseline and weeks 1, 2, 4, 6, 8, 10, 12, 16, 20, 24, and 28, with weeks 12–28 designated as the continuation phase of the study. Measurement-based care provided rigorous dosing at each visit (
37–
39); dosage adjustments were based on the 16-item Quick Inventory of Depressive Symptomatology-Clinician-rated (QIDS-C-16) (
26,
27,
40), which was extracted from the IDS-C-30 (
27), and the Frequency, Intensity, and Burden of Side Effects Rating (FIBSER) (
41) and was guided by the CO-MED Operations Manual (available at
www.co-med.org).
Treatment was randomly assigned and stratified by clinical site with a Web-based randomization system (
42). Random block sizes of three and six minimized the probability of identifying the next treatment assignment. Dosing schedules were based on prior reports (
43–
45), and doses were increased only in the context of acceptable side effects. Dose changes were made by the treating physician in collaboration with the clinical research coordinator after review of the data collected at the visit. Participants could exit the study if unacceptable or intolerable side effects occurred that could not be resolved with dose reduction or medication treatment of side effects.
The dosing protocols are described below.
Ecitalopram plus placebo.
Escitalopram began at one tablet (10 mg per day) and increased to two tablets (20 mg per day) at week 4 (if QIDS-C-16 score was >5). Pill placebo (one pill) was started at week 2, with the option to increase to two pills at week 4 if the QIDS-C-16 score was >5 and side effects were tolerable.
Bupropion-sustained release (SR) plus escitalopram.
Bupropion-SR (150 mg per day) was started at baseline and increased to 300 mg per day at week 1. Escitalopram began at 10 mg per day at week 2. At week 4, bupropion-SR could be raised to 400 mg per day and escitalopram could be increased to 20 mg per day if the QIDS-C-16 score was >5 and side effects were tolerable. At week 6 and beyond, if not already done, the bupropion-SR dose was increased to a maximum of 400 mg per day (200 mg per day b.i.d.) and escitalopram to 20 mg per day if the QIDS-C-16 score was >5 and side effects were tolerable to the patient.
Venlafaxine-extended release (XR) plus mirtazapine.
Venlafaxine-XR was begun at 37.5 mg per day for three days and then raised to 75 mg per day. At week 1, venlafaxine-XR was raised to 150 mg per day. At week 2 if the QIDS-C-16 score was >5, mirtazapine was added (15 mg per day). At week 4, if the QIDS-C-16 score was >5, venlafaxine-XR could be raised to 225 mg per day and mirtazapine could be increased to 30 mg per day. At week 6, if the QIDS-C-16 score was >5, mirtazapine could be raised to 45 mg per day (maximum dose). At week 8, if the QIDS-C-16 score was >5, venlafaxine-XR could be raised to 300 mg per day (maximum dose).
Concurrent treatments.
Only protocol antidepressant medications were allowed. Other treatments with possible antidepressant effects were proscribed, including depression-targeted, empirically validated psychotherapies for depression. Other therapies (such as supportive therapy and couples therapy) were allowed, as were medications for general medical conditions. Given the bupropion-SR inhibition of the 2D6 isoenzyme, clinicians were alerted to recognize nonprotocol medications (such as type 1C antiarrythmics and beta blockers) for which serum levels or dose adjustments might be needed. Medications to treat antidepressant side effects were allowed to mimic practice and enhance retention.
Research outcomes
Assessments were collected at baseline and at all subsequent visits. The primary outcome, symptom remission, was based on the self-report version of the 16-item QIDS (QIDS-SR-16) (
26,
27,
40). Remission was determined on the basis of the last two consecutive measurements obtained during the acute trial to ensure that a single “good week” was not falsely signaling remission. At least one of these ratings had to be <8, and the other had to be <6. If participants exited before 12 weeks, their last two consecutive QIDS-SR-16 scores were used to assess remission. Those exiting before having two postbaseline measures were considered as not reaching remission.
Participants could exit the study if they had received a maximally tolerated dose for at least four weeks by week 8 without receiving a ≥30% reduction from baseline QIDS-C-16. They could enter continuation treatment if they had received an acceptable benefit (QIDS-C-16 score ≤9 by week 12) or if they reached a QIDS-C-16 score of 10–13 with the clinician and participant judging the benefit to be substantial enough to indicate treatment continuation.
Secondary outcomes included attrition; response (QIDS-SR-16 score reduction of ≥50%); anxiety, measured by the anxiety subscale of IDS-C-30 (
26–
29); function, measured by the Work Productivity and Activity Impairment scale (
46) and the Work and Social Adjustment Scale (WSAS) (
47); quality of life, measured by the Quality of Life Inventory (QOLI) (
48,
49); side-effect burden, measured by the FIBSER (
41); and specific side effects, measured by the Systematic Assessment of Treatment Emergent Events-Systematic Inquiry (SAFTEE-SI) (
50,
51).
Statistical analyses
Descriptive statistics, including measures of central tendency and dispersion, were computed for continuous data. Frequency distributions were estimated for categorical data. The appropriate parametric (t test) or nonparametric (chi square and Wilcoxon tests) test was used to ensure a balanced distribution of the sociodemographic, psychiatric, and medical characteristics among the groups.
At weeks 12 and 28, unadjusted and adjusted outcomes among the racial-ethnic groups were compared via regression models. The type of regression models varied by outcome and included linear regression, logistic regression, ordinal logistic regression, and negative binomial regression models. Potential confounders were identified with a stepwise logistic regression model with race-ethnicity as the outcome and all other baseline characteristics as independent variables. Variables that remained in the final stepwise model were considered as potential confounders in the adjusted models. The moderating effect of race-ethnicity on treatment was evaluated on two outcomes, severity of depression (QIDS-SR-16) and side effect burden (FIBSER burden subscale), at 12 and 28 weeks. For severity of depression, a linear regression model was fit, and for side effect burden an ordinal logistic regression model was fit. Both models included main effects for treatment and race-ethnicity, as well as the two-way interaction between treatment and race-ethnicity. All analyses were considered to be exploratory, and a type I error or a p value <.05 was used as a threshold to identify statistical significance. When three-group comparisons were made, the p value for significance was adjusted to p<.017. No adjustments were made for multiple comparisons, so results should be interpreted accordingly.
Results
Of 835 participants invited to consent to be screened for the study, 734 (88%) signed consent; of those, 665 (91%) were eligible and were randomly assigned to a treatment group.
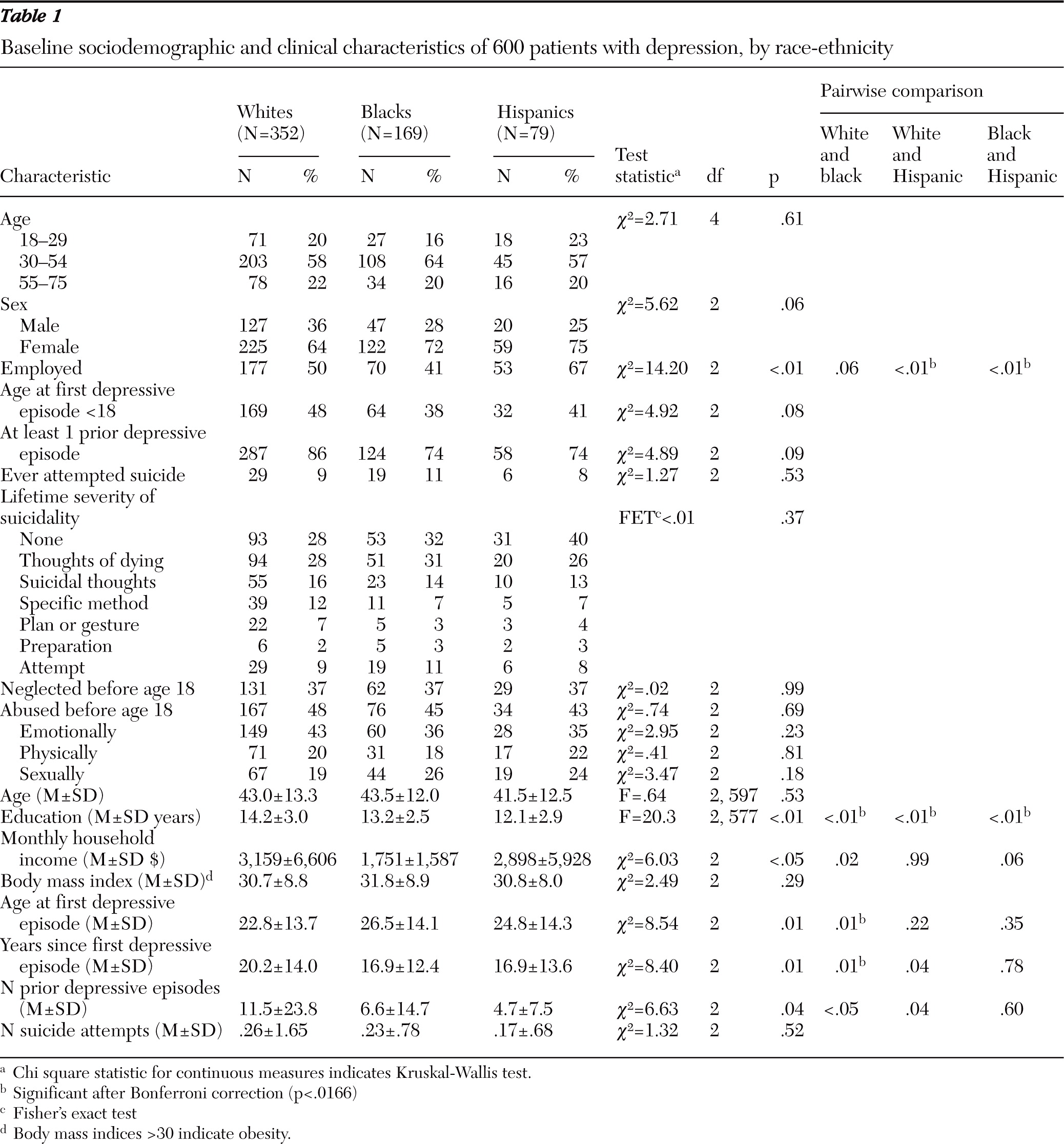
Available data on race and ethnicity indicated that the enrolled sample consisted of 431 whites, 174 blacks, 22 Asian American or Pacific Islanders, seven Native Americans, and nine participants who endorsed more than one group. Because of the small numbers in the latter groups, we restricted comparisons to 352 non-Hispanic whites (59%), 169 blacks (28%), and 79 white Hispanics (13%). As shown in
Table 1, Hispanics had a significantly higher rate of employment (67%) compared with whites (50%, p<.01) and blacks (41%, p<.01). Blacks had significantly more years of education than Hispanics (mean of 13.2 years versus 12.1 years, p<.01), and whites had significantly more years of education than both of these groups (14.2 years, p<.01). Whites had the earliest age of first major depressive episode (mean age=22.8), which was significantly earlier than for blacks (mean age=26.5, p<.01) (
Table 1).
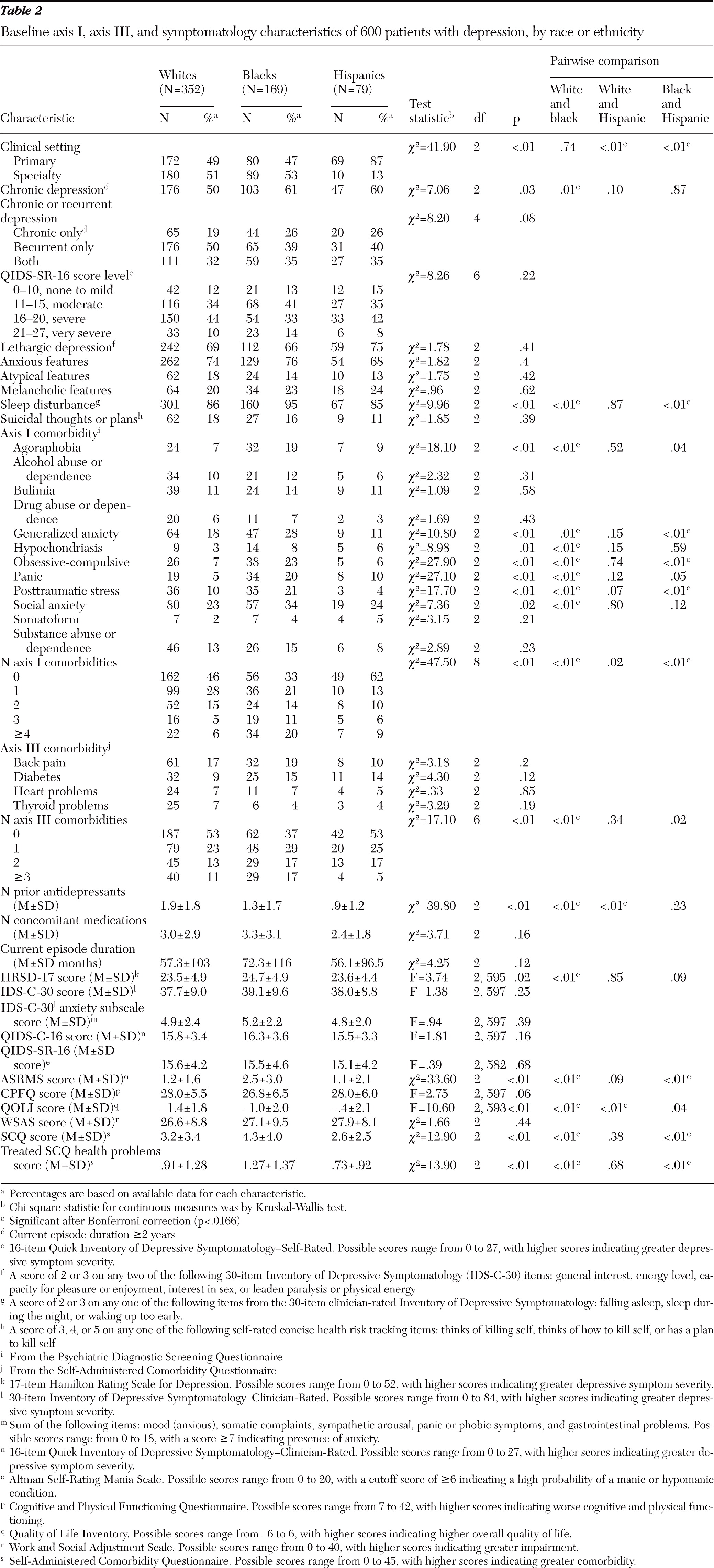
Compared with whites (50%), a significantly greater percentage of blacks (61%, p=.01) had chronic depression; there was no difference between blacks and Hispanics (60%). Hispanics were more likely to be seen in primary care settings (87%) compared with whites (49%, p<.01) or blacks (47%, p<.01) (
Table 2).
On some measures, blacks had the highest levels of psychiatric and medical comorbidity. Compared with whites and Hispanics, a greater percentage of blacks had agoraphobia, generalized anxiety disorder, panic disorder, obsessive-compulsive disorder, posttraumatic stress disorder, and social anxiety disorder (PDSQ), although anxious features did not significantly differ on the HRSD-17 or IDS-C-30. Depressive symptom severity did not differ at a clinically important level across groups. Black participants scored higher on the ASRMS (mean=2.5 out of a possible 20) than both whites (1.2, p<.01) and Hispanics (1.1, p<.01), but all scores were low and not indicative of manic behavior. On the QOLI, whites reported the poorest quality of life (mean score=−1.4 out of a possible 6), followed by blacks (−1.0, p<.01) and Hispanics (−.4, p<.01) (
Table 2).
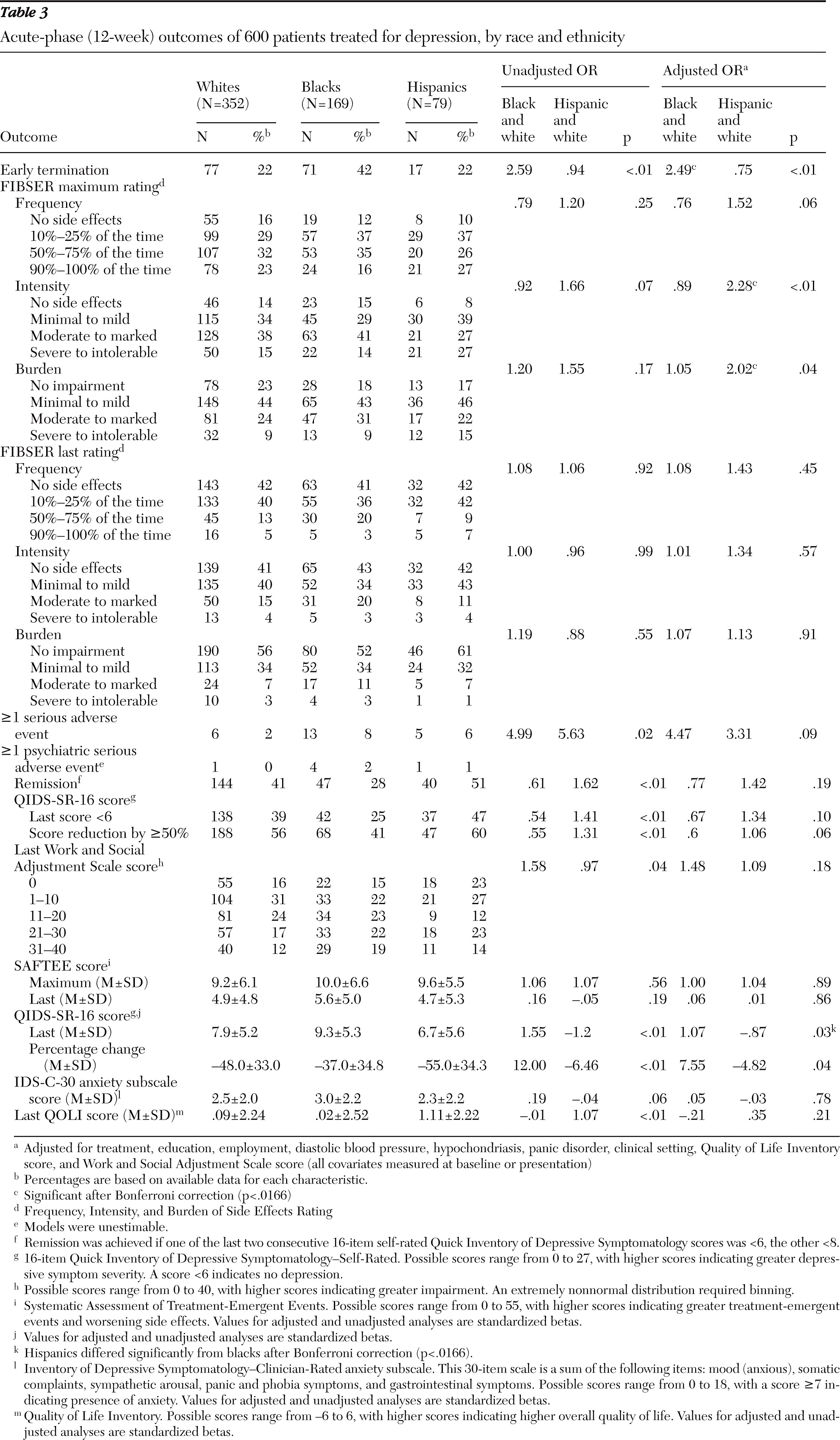
A greater proportion of black participants exited the study early compared with whites and Hispanics (
Table 3). Approximately 20% exited by week 4 (versus about 11% of other groups); 27% exited by week 6 (versus about 14% of other groups), and 42% exited before completing week 12 (versus about 21% of whites and 26% of Hispanics). The odds ratio (OR) for blacks to exit early was about 2.5 compared with whites. This same pattern of early termination by blacks was seen in continuation treatment (
Table 4). In general, final doses received were similar, although blacks less often received the highest dose of bupropion-SR and escitalopram (data not shown), likely because of their early exit. In the acute phase, Hispanic participants had a higher maximum intensity (OR=2.28) and burden of side effects (OR=2.02) compared with whites (
Table 3). Side effects during continuation treatment were equal across groups (
Tables 3 and
4). After 12 weeks and before adjustment for baseline differences, Hispanics had the highest remission (51%) and response (60%) rates and blacks the lowest (28% and 41%, respectively), and Hispanics had a significantly lower last QIDS-SR-16 score (6.7 out of a possible 40) than blacks (9.3), indicating less severe depression. After adjustment for baseline differences, the only significant difference at week 12 was that compared with blacks, Hispanics had a lower last QIDS-SR-16 score, and there were no significant differences in these three outcomes across groups at week 28 (
Table 4). There were no significant differences regarding effect of treatment among the racial-ethnic groups at weeks 12 and 28 (
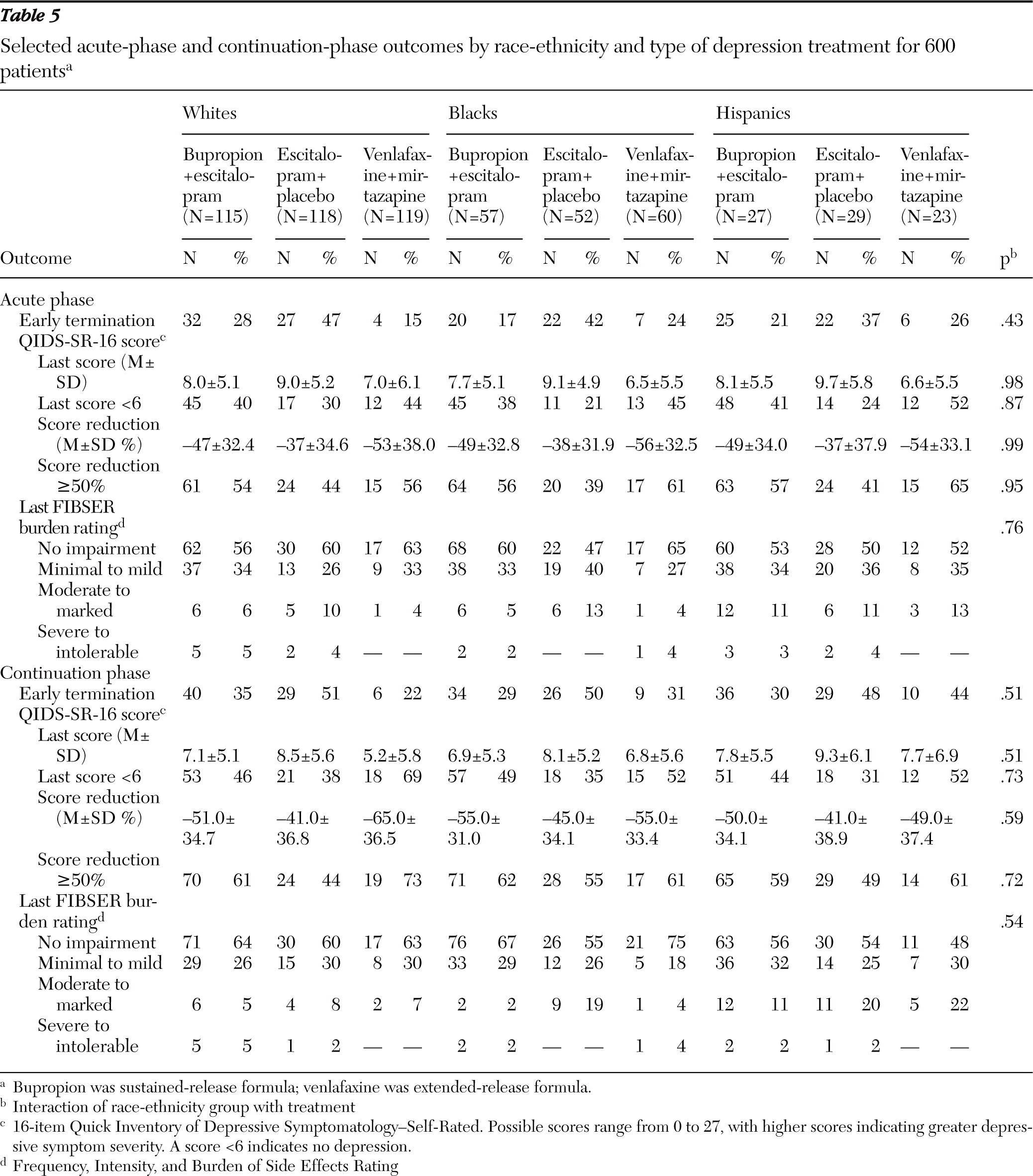
Table 5).
Discussion
As in the STAR*D study, this study showed considerable socioeconomic and demographic baseline differences among racial-ethnic groups (
14). Whether this represents an accurate picture of those seeking treatment in the general population or is related to the participating sites is unclear. Blacks had more general medical and perhaps psychiatric comorbidity, and both blacks and Hispanics had higher rates of chronic depression compared with whites, although the comparison between Hispanics and whites was not statistically significant. These differences likely represent socioeconomic realities and poorer access to care or greater reluctance to seek care for members of minority groups, perhaps leading to increased chronicity and comorbidity (
1–
7). Despite these differences, baseline severity of depression was similar across groups.
Although more Hispanics were recruited from primary care sites (perhaps reflecting help-seeking behavior), outcomes for Hispanics were equal to (or better than) those in the other groups, similar to the findings of STAR*D and other studies using measurement-based care (
14,
52). In the unadjusted analyses at 12 weeks, blacks fared worst and Hispanics best in achieving remission. Once adjustments were made for baseline differences, outcomes were similar among groups.
There were few differences in side effects among the groups and no differences in adverse events. Compared with whites, Hispanics were more likely to have higher scores for maximum intensity and burden of side effects during the acute phase but not the continuation phase. Despite this, Hispanic participants did not terminate treatment earlier than participants in the other groups, suggesting that the side effect load was tolerable. Despite putative differences in pharmacogenetics in racial-ethnic populations and their effects on treatment response (
53–
56), we saw no significant differential pattern of side effects or outcome across groups that could be attributed to genetic differences.
Blacks exited the acute and continuation phases at higher rates than both of the other groups. This may account for their poorer remission and response rates, because they may have exited treatment before benefiting from treatment, and those who dropped out very early were considered nonremitters. Blacks had more comorbid anxiety disorders, according to PDSQ score, but baseline anxiety symptoms, as measured by the HDRS-17 and the IDS-C-30 anxiety subscales, did not differ among groups. When additional analyses controlled for the presence of generalized anxiety disorder, outcomes did not significantly differ. Perhaps the PDSQ, a self-rated scale that approximates psychiatric diagnoses, yielded a different pattern of response than the two clinician-rated scales—an area of study in which cultural factors may have a part and that may deserve further scrutiny. Black participants were also more likely to be unemployed and have lower annual incomes—both factors linked to attrition in prior studies (
16,
57).
It has been reported that for blacks in particular, and to some degree for Hispanics, antidepressant medication may be a less acceptable treatment for depression compared with psychosocial treatments (
58) and that medication adherence may be poorer among patients from racial-ethnic minority groups (
59). These could be additional factors contributing to the early exit of blacks from this study. Previous depression research studies have reported higher dropout rates for blacks (
14,
16) in community samples of participants with mental disorders (
59,
60) and in studies of other chronic diseases (
61–
63). This presents a major challenge to clinicians, and new strategies must be devised to address this issue. Perhaps eliciting patient concerns about enrolling in medication trials and providing more specific education about these concerns would enhance adherence and decrease dropout.
This study demonstrated that, after adjustment for baseline differences in sociodemographic characteristics and comorbidity, antidepressant medication outcomes across racial-ethnic groups were similar regardless of whether a single agent or two agents were used. This finding confirms and extends previous findings (
10,
11,
14,
15) that when dosing of medications based on monitoring of symptoms and side effects is performed, there are no differences in outcome across racial-ethnic groups. Furthermore, there was no indication that a particular medication combination was more effective in any racial-ethnic group. One might argue that blacks, who on average had lower maximum doses of escitalopram and bupropion-SR, may have received medication at too low a dose. However, this “undermedication” was likely accounted for by higher rates of early termination in this group.
Several limitations of the study should be considered. Our strategy was to have participants self-identify their background, but these groupings do not imply sociocultural or genetic homogeneity, and it is likely that there was substantial heterogeneity in all three study groups. Assigning participants to the categorical dimensions of race or ethnicity is problematic, with no acceptable gold standard (
64). The sample size in the groups was relatively modest, although it was larger than that in many outcome studies of this type. CO-MED did not use a placebo group, so we cannot say for certain how the outcomes of the treatment groups would compare with those from no active treatment. Finally, enrollees had chronic or recurrent major depressive disorder, and thus we cannot generalize the results to patients with less chronic and single-episode major depressive disorder.
Conclusions
After adjustment for baseline sociodemographic and comorbidity differences, outcomes for participants receiving single or combination antidepressant treatment were similar for white, black, and Hispanic participants. This study adds to the literature showing that disparities in outcome among participants from different racial-ethnic groups can be minimized with measurement-based care.
Acknowledgments and disclosures
This project was funded by the National Institute of Mental Health (NIMH) under contract N01MH90003 to the University of Texas Southwestern Medical Center at Dallas. The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services, nor does mention of commercial products or organizations imply endorsement by the U.S. government. The NIMH had no role in the drafting or review of the manuscript, nor in the collection or analysis of the data. The authors appreciate the support of Forest Pharmaceuticals Inc., GlaxoSmithKline, Organon Inc., and Wyeth Pharmaceuticals in providing medications at no cost for this trial. The authors also acknowledge the editorial support of Jon Kilner, M.S., M.A.
Dr. Lesser reports no competing interests. Dr. Zisook has received research support from Pamlab, LLC. Dr. Gaynes has received grant support from M-3 Corporation. Dr. Wisniewski has been a consultant for Dey Pharmaceuticals and Venebio. Mr. Luther reports no competing interests. Dr. Fava has received research support from or been an advisor to the following: Alkermes, Inc.; AstraZeneca; BioResearch; BrainCells Inc.; Bristol-Myers Squibb; CeNeRx BioPharma; Clinical Trials Solutions, LLC; Clintara, LLC; CNS Response, Inc.; Covance; Covidien; Cypress Pharmaceutical, Inc.; Dinippon Sumitomo Pharma Co. Inc.; Eisai Inc.; Eli Lilly and Company; EnVivo Pharmaceuticals, Inc.; Euthymics Bioscience, Inc.; Forest Pharmaceuticals, Inc.; Ganeden Biotech, Inc.; GenOmind, LLC; GlaxoSmithKline; Grunenthal GmbH; Icon Clinical Research; i3 Innovus/Ingenix; Janssen Pharmaceutica; Johnson & Johnson PRD; Lundbeck Inc.; MSI Methylation Sciences, Inc.; Naurex, Inc.; NextWave Pharmaceuticals; Novartis AG; Otsuka Pharmaceuticals; Pamlab, LLC.; Pfizer Inc.; PharmoRx Therapeutics; Photothera; Puretech Ventures; PsychoGenics; Psylin Neurosciences, Inc.; Rexahn Pharmaceuticals, Inc.; Ridge Diagnostics, Inc.; Roche Pharmaceuticals; RCT Logic, LLC; Sanofi-Aventis US LLC; Servier Laboratories; Sunovion Pharmaceuticals; Supernus Pharmaceuticals, Inc.; Takeda Pharmaceutical Company Ltd; Tal Medical, Inc.; and Vanda Pharmaceuticals, Inc. He has equity holdings in Compellis. He has been a speaker for or published with the Belvoir Media Group; CME Institute/Physicians Postgraduate Press, Inc.; and the MGH Psychiatry Academy. In addition, Dr. Fava receives royalties or patent or other income as follows: patent for Sequential Parallel Comparison Design (SPCD); patent for research and licensing of SPCD with RCT Logic; patent application for a combination of azapirones and bupropion in major depressive disorder; copyright royalties for the MGH Cognitive and Physical Functioning Questionnaire, Sexual Functioning Inventory, Antidepressant Treatment Response Questionnaire, Discontinuation-Emergent Signs and Symptoms, and SAFER; and royalties from Lippincott, Williams & Wilkins and World Scientific Publishing Co. Dr. Khan is on the speaker panel for Merck Schering-Plough. Dr. McGrath receives research and grant support from Roche Pharmaceuticals. Dr. Warden reports no competing interests. Dr. Rush has received royalty payments from Guilford Press, has been a consultant to Brain Resource, Inc., and has received a travel grant from CINP and a speaking fee from Singapore College of Family Physicians. Dr. Trivedi has been a speaker or received consultant honoraria from Abbott Laboratories, Inc.; Alkermes; AstraZeneca; Axon Advisors; Bristol-Myers Squibb; Cephalon, Inc.; CME Institute of Physicians; Eli Lilly and Company; Evotec; Forest Pharmaceuticals; GlaxoSmithKline; Johnson & Johnson PRD; Lundbeck, Inc.; MedAvante; Neuronetics; Otsuka Pharmaceuticals; Pamlab; Pfizer, Inc.; PgxHealth; Rexahn Pharmaceuticals; SHIRE Development; Takeda Pharmaceutical Company Ltd.; Tal Medical/Puretech Venture; and Transcept. Dr. Trivedi has also received research support from Naurex, Inc.; Targacept; and Valient.