Serotonin neuronal dysfunction is implicated in several substance abuse and clinical disorders, including alcoholism (
1), depression (
2), suicidality (
3), and impulsivity (
4). Increased imipramine binding to the serotonin transporter has been detected in hippocampal samples from deceased subjects with evidence of ethanol exposure (
5) and considerable evidence suggests that changes occur in platelet serotonin transporter function in alcoholics (
6–
8). The neurochemical and clinical implications of these findings remain uncertain, but recent evidence indicates that alterations in monoamine transporter function can significantly modulate overall neuronal function (
9,
10).
Cocaine acutely blocks the uptake of both serotonin and dopamine by binding to the transporters specific for each neurotransmitter. Although the effect of cocaine on dopamine uptake is critical for reward, animal experiments suggest that acute blockade of serotonin transporter function may also contribute to clinical symptoms such as euphoria, anxiety, and craving (
11,
12). Chronic cocaine exposure increases binding sites on the dopamine transporter in the striatum of human users (
13,
14), perhaps as a compensation for chronically diminished function. This effect might similarly occur with the serotonin transporter.
In the present investigation, we tested the hypothesis that alterations in brain serotonin transporter exist in chronic users of ethanol and cocaine. Serotonin transporter binding sites and serotonin transporter mRNA levels were quantified in postmortem samples from several groups of human subjects who chronically abused ethanol, cocaine, or both. Serotonin transporter binding sites were quantitated by radioligand autoradiography and serotonin transporter mRNA was quantitated by in situ hybridization in midbrain samples containing dorsal and median raphe nuclei (the location of serotonin cell bodies that innervate the forebrain). Serotonin transporter promoter genotype was also determined for each subject.
RESULTS
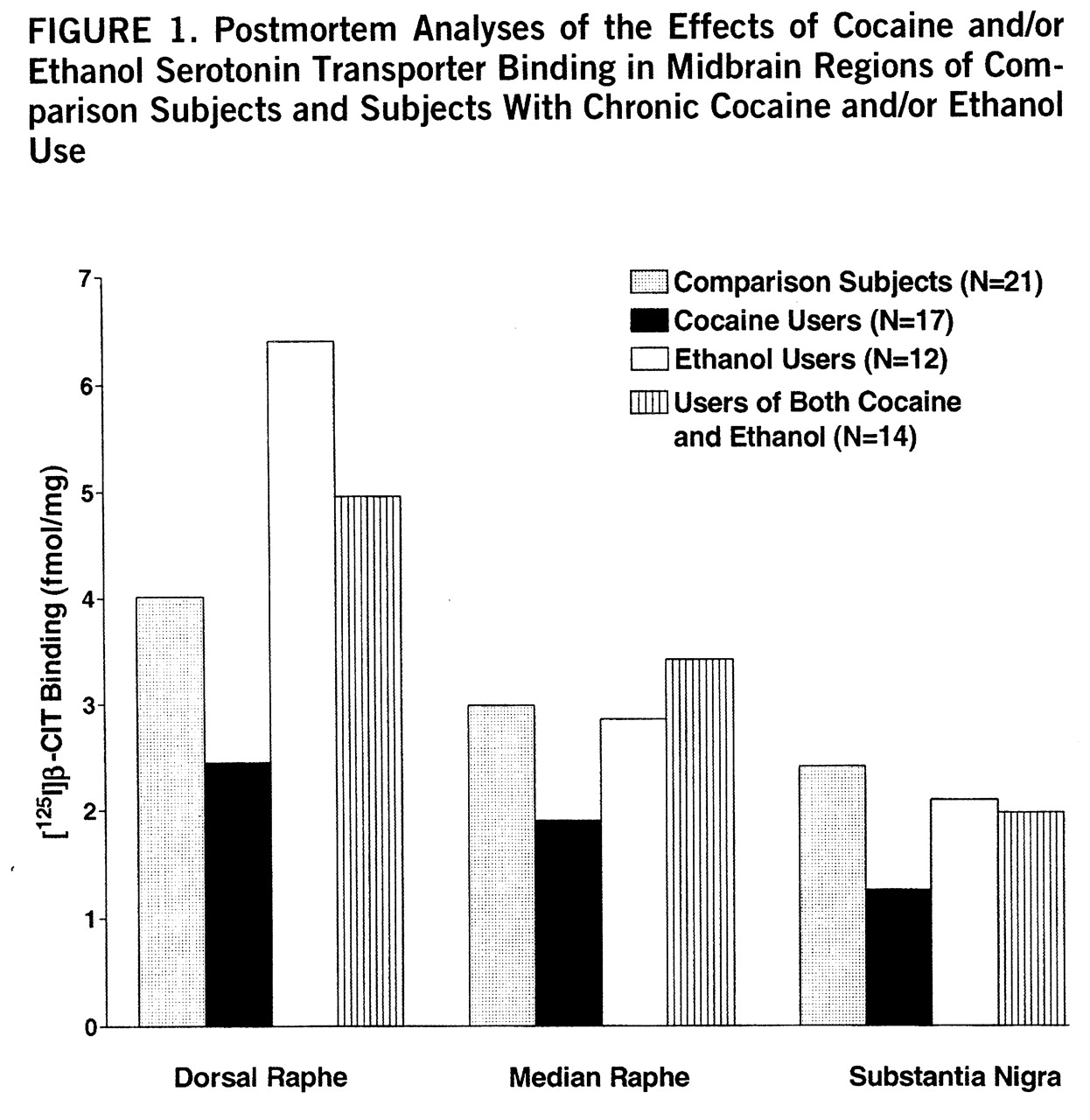
Figure 1 (nontransformed values) displays the overall effects of cocaine and ethanol on binding in the three midbrain regions. Significant cocaine-by-ethanol-by-genotype interactions were found in the initial MANOVA analysis by using the raw or log-transformed data (Wilks's lambda=0.86, F=2.66, df=3,49, p=0.03). [
125I]β-CIT binding in cocaine users appeared to be lower in all three regions, but reached statistical significance only in the dorsal raphe: dorsal raphe (mean=4.0 fmol/mg, SD=0.6, for comparison subjects, versus mean=2.1 fmol/mg, SD=0.3, for cocaine users) (t=2.51, df=32, p=0.02); median raphe (mean=2.9 fmol/mg, SD=0.6, for comparison subjects, versus mean=1.9 fmol/mg, SD=0.3, for cocaine users) (t=1.76, df=32, p=0.09); and substantia nigra (mean=2.3 fmol/mg, SD=0.5, for comparison subjects, versus mean=1.2 fmol/mg, SD=0.2, for cocaine users) (t=1.80, df=32, p=0.08). The apparent difference between comparison subjects and ethanol users in dorsal raphe was not statistically significant (mean=4.0 fmol/mg, SD=0.6, for comparison subjects, versus mean=6.4 fmol/mg, SD=1.6, for the ethanol users) (t=1.63, df=27, p=0.11), possibly because variability was high and the effect was influenced by genotype. In contrast to serotonin transporter binding sites, there were no drug abuse effects on serotonin transporter mRNA levels.
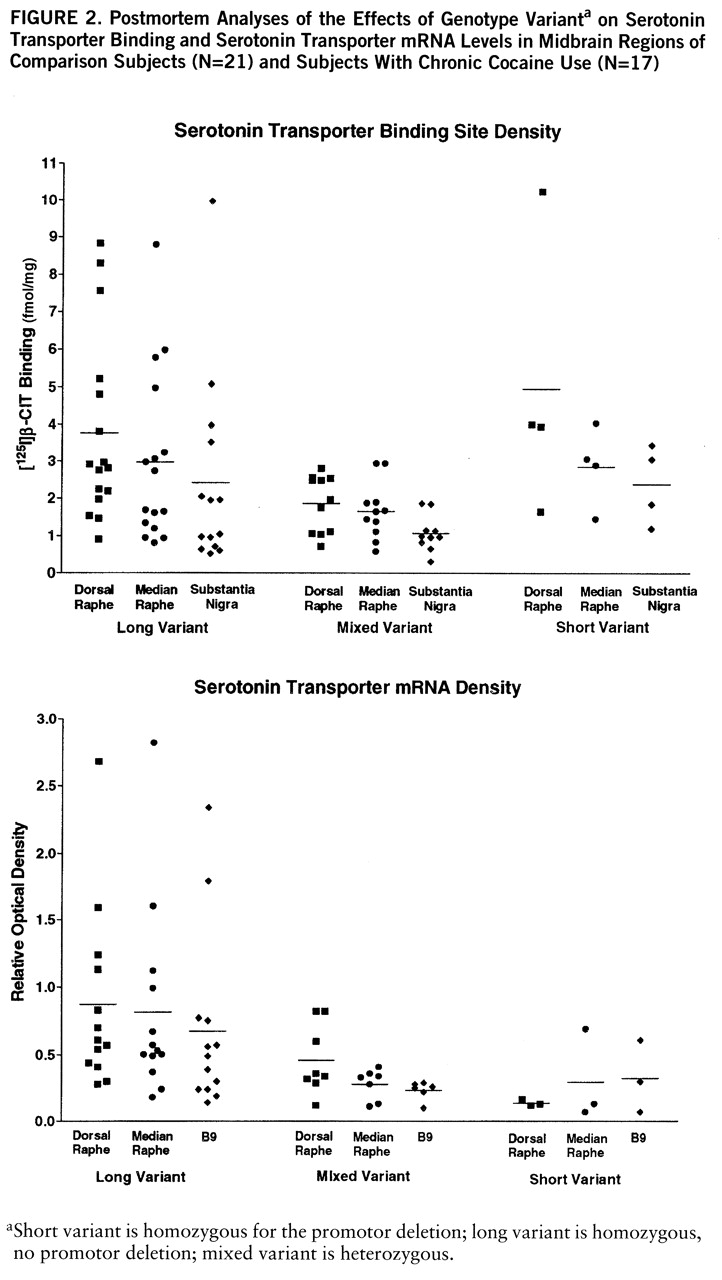
There was a consistent genotype effect on both serotonin transporter binding and mRNA levels across regions (
figure 2). For this part of the study, the ethanol users were excluded because they displayed a genotype-by-ethanol interaction. Serotonin transporter binding varied significantly by genotype and region (two-way ANOVA, genotype-by-region, genotype main effect, F=8.02, df=1,87, p<0.001; region, F=5.75, df=1,87, p=0.005, no significant interaction). Serotonin transporter binding in subjects with the heterozygous genotype was clearly lower than in subjects with the long genotype, while subjects with the short genotype appeared equivalent to long genotype, perhaps because of the small number of subjects.
Serotonin transporter mRNA levels also varied significantly by genotype, but not by region (two-way ANOVA, genotype-by-region, genotype main effect, F=8.28, df=1,67, p<0.001; region, F=1.64, df=1,67, n.s.). Serotonin transporter mRNA levels in subjects with the mixed and short genotypes were clearly lower than in subjects with the long genotype.
There was a statistically significant ethanol-by-genotype interaction found by MANOVA which was limited to the dorsal raphe in the midbrain areas examined for this study. [125I]CIT binding in subjects with either the short or mixed genotype was significantly higher in ethanol-using subjects than in comparison subjects (mean=7.2 fmol/mg, SD=1.5, versus mean=2.7 fmol/mg, SD=0.6) (t=2.97, df=25, p=0.006, two-tailed). Values were similar in ethanol users and comparison subjects with the long variant (mean=3.5 fmol/mg, SD=0.5, versus mean=3.6 fmol/mg, SD=0.6) (n.s.). In subjects without ethanol diagnoses, 8% (N=4) had the short genotype; 17% (N=2) of the ethanol-user subjects had the short genotype. Inspection of the clinical data on the short- versus long-genotype ethanol users did not reveal any distinguishing features. Several individuals were diagnosed with antisocial personality in each group and Hamilton scores were similar for those individuals who scored any points. There were no cocaine interactions with genotype, nor any interactive cocaine effects on mRNA levels.
Genotype frequencies for the four groups were as follows: comparison subjects—three unable to amplify, three short, six mixed, nine long; ethanol-using subjects—two short, five mixed, five long; cocaine-using subjects—one short, seven mixed, nine long; and for subjects who were users of both cocaine and ethanol—no short, six mixed, eight long. The frequency of the long allele ([(homozygous long N×2)+mixed N] divided by (total N×2), where N is the number of subjects) did not differ significantly among the subject groups: comparison subjects, 67% ethanol-using subjects, 62% cocaine-using subjects, 73% and subjects who were users of both cocaine and ethanol, 79% (χ2=1.87, df=3, p=0.60). However, our total numbers were small and our case-control study design is imperfect for testing for possible association between genotype and clinical predilection to either cocaine or ethanol abuse.
Binding and mRNA levels were not significantly correlated with each other in the same regions within individuals of the same group. There were no significant differences in age or postmortem interval among the various groups, and neither binding nor mRNA levels correlated with these parameters. No significant race, socioeconomic, or sex differences were discovered. Neither binding nor mRNA levels were different in subjects with mood disorder or psychotic diagnoses, or in subjects with opioid abuse diagnosis, but the statistical power for these analyses was limited.
DISCUSSION
The present findings are consistent with one earlier postmortem human study (
5), which found increased [
3H]imipramine binding in hippocampal dentate and pyramidal cell layers from subjects who had died after acute ethanol exposure. Animal experiments in alcohol-preferring strains of rats have reported diffusely increased binding to the serotonin transporter, although not all studies found this effect (
29,
30). One report found no basal differences between strains, but noted that ethanol exposure induced increased serotonin transporter binding in several brain regions (
31). Together, these earlier experiments suggest that ethanol use or a predisposition to use may involve brain serotonin transporter.
No previous studies have explored cocaine effects in postmortem human brain. Cunningham et al. (
32) reported increased [
3H]imipramine binding in medial prefrontal cortex, frontal cortex, sulcal prefrontal cortex, and dorsal raphe nuclei of rats treated with cocaine (15 mg i.p. b.i.d. for 7 days). In addition, Belej et al. (
33) reported that oral self-administration of cocaine in rats increased [
3H]paroxetine binding in three of 53 regions, including by 21% in the accumbens. These rat studies, in view of our results, tentatively suggest that cocaine effects on the serotonin transporter may be different in humans; however, dosing amounts and schedules may also contribute to differing results. This is clearly the case with cocaine's effect on the dopamine transporter (
13,
34,
35).
Several anatomical issues complicate interpretation of the present results. Serotonin transporter function in the substantia nigra and the two raphe nuclei may have distinct overall effects on serotonergic signaling. In the substantia nigra, serotonin transporter molecules are axonally located and might influence postsynaptic signaling by modulating synaptic serotonin concentrations. In contrast, serotonin transporter molecules in the raphe are somatodendritic and represent collateral innervation from proximal and distal serotonergic neurons. Somatodendritic transporters might be expected to contribute to inhibition and synchronization of cell firing rates. Because of this, an increase in somatodendritic serotonin transporter function, suggested by increased dorsal raphe [125I]CIT binding in alcoholics with the short genotype, might serve to increase serotonin firing rates by diminishing self-inhibition. Conversely, in cocaine-using subjects, a decrease in dorsal raphe serotonin transporter activity might decrease serotonergic firing, through increased self-inhibition, while a similar decrease in serotonin uptake in the substantia nigra would tend to promote serotonergic signaling by delaying the clearance of synaptic serotonin. To better reconcile these issues considerably more information is needed about the status of other perisynaptic factors also critically involved, especially release rates and pre- and postsynaptic receptor sensitivities.
The data from this study represent the second reciprocal relationship we have detected between dopamine and serotonin parameters in cocaine users. In earlier experiments, we detected increased striatal dopamine transporter binding, and in a different group of subjects, higher frontal cortex serotonin and lower dopamine levels, than in comparison subjects (
36). In the future it would be desirable to measure both monoamine transporter binding levels and neurotransmitter levels from the same groups. A useful feature of postmortem studies is the possibility of examining a number of parameters in samples from contiguous sections in the same individual.
The present genotype results appear generally consistent with those of Lesch et al. (
15), who examined binding, mRNA levels, and uptake in lymphoblasts from nine individuals. Lesch et al. found that heterozygote genotypes were best classified with the short variants, which was also the case for serotonin transporter mRNA levels (but not serotonin transporter binding) in the present study. However, the apparently elevated binding among those subjects with the short variant was influenced by one very high value, and might simply reflect too small a sample. It is also possible, however, that there is a true difference between heterozygotes and either type of homozygote. Unlike Lesch et al., we were not able to examine actual serotonin uptake, which we have found is not satisfactorily recoverable in frozen samples because of variable postmortem degradation.
The serotonin transporter polymorphism may be important clinically and one can anticipate an interest in further screening of alcoholic patients and depressed patients, particularly those refractory to treatment, as well as screening of other populations suspected of serotonin transporter abnormalities, such as those with autistic disorder (
37). Both genotype and peripheral serotonin transporter binding can be fairly easily explored in peripheral tissue, although it is possible that critical adaptations only occur in certain brain regions.
The detection of genotype, ethanol, and cocaine effects in this relatively small and complex population suggests that each of the effects may be fairly robust. Fortuitously, cocaine and ethanol alterations were in opposite directions, allowing us to distinguish their differing effects. However, the size of these groups was rather small and caution should be exercised in extending these findings until further replication occurs. Confidence in the findings would be increased if the number of subjects had been larger, more extensive sampling were done in these regions, and other brain regions were examined. Also, our clinical classification may contain some errors because of the nature of the clinical data. In particular, it may be that ethanol dependence could have been missed in some cocaine users, although efforts were made to prevent this. An ideal reclassification might actually increase the apparent cocaine effect since there were some high values in subjects using only cocaine who might have been more accurately included in the users of both cocaine and ethanol category.
In conclusion, it appears that regulation of serotonin transporter binding sites is a dynamic process, with distinct region- and substance-specific patterns that are more than local responses to functional blockade. There may be a reciprocal relationship between cocaine and ethanol effects in the dorsal raphe, perhaps clinically important for patients that are dually dependent. In sum, the present data suggest that a better understanding is needed of 1) the time course and doses involved in cocaine- and ethanol-induced changes; 2) the range of brain regions involved in cocaine, ethanol, and genotype effects; and 3) the clinical features associated with cocaine- and ethanol-regulated serotonin transporter binding, and various serotonin transporter genotypes.