Tardive dyskinesia is a movement disorder that affects 20%–40% or more of patients treated chronically with neuroleptic drugs
(1). The manifestations of tardive dyskinesia may include adventitious movements of the oral-facial region, choreoathetosis of the extremities, and lordotic posturing. Although the theory that striatal postsynaptic dopamine receptor supersensitivity causes tardive dyskinesia has been widely accepted for two decades, there is evidence that challenges this model
(2). Although acute administration of neuroleptics temporarily increases the firing of dopamine neurons, chronic treatment with neuroleptics leads to a decrease in their firing rate caused by depolarization block
(3)). Dopamine synthesis and release decrease after an acute elevation of dopamine turnover with haloperidol treatment; dopamine metabolite levels return to normal with chronic treatment
(4). Notably, striatal dopamine metabolites are reduced in monkeys with dyskinesia caused by long-term neuroleptic treatment. Finally, postmortem neurochemical studies have not revealed a correlation between dopamine receptor up-regulation and tardive dyskinesia
(5). Thus, an excessive activation of postsynaptic striatal dopamine receptors is not consistent with the time course or persistence of tardive dyskinesia.
An alternative hypothesis supports a neurodegenerative process affecting striatal efferents analogous to the process in Huntington’s disease. Tardive dyskinesia is similar to Huntington’s disease in that both diseases present with choreoathetoid movements and pathological changes in the striatum. Christensen et al.
(6) reported neuronal loss in a postmortem brain study of the basal ganglia of patients with persistent tardive dyskinesia, and similar losses have been described in rats treated chronically with neuroleptics
(7-
9). Importantly, loss of the presynaptic markers for the striatal-pallidal and nigral γ-aminobutyric acid (GABA), GABAergic neurons, and glutamic acid decarboxylase have been observed in a primate model for tardive dyskinesia and in postmortem studies of patients with tardive dyskinesia
(10-
12).
Presynaptic dopamine D
2 receptors inhibit the release of glutamate from excitatory cortical striatal projections
(13). Therefore, neuroleptic blockade of these receptors increases the synaptic release of aspartate and glutamate in the striatum
(14,
15). Persistent activation of glutamate ionotropic receptors has long been known to cause neuronal degeneration
(16). Oxidative damage mediates the delayed neuronal degeneration caused by activation of
N-methyl-D-aspartic acid (NMDA) and non-NMDA glutamate ionotropic receptors
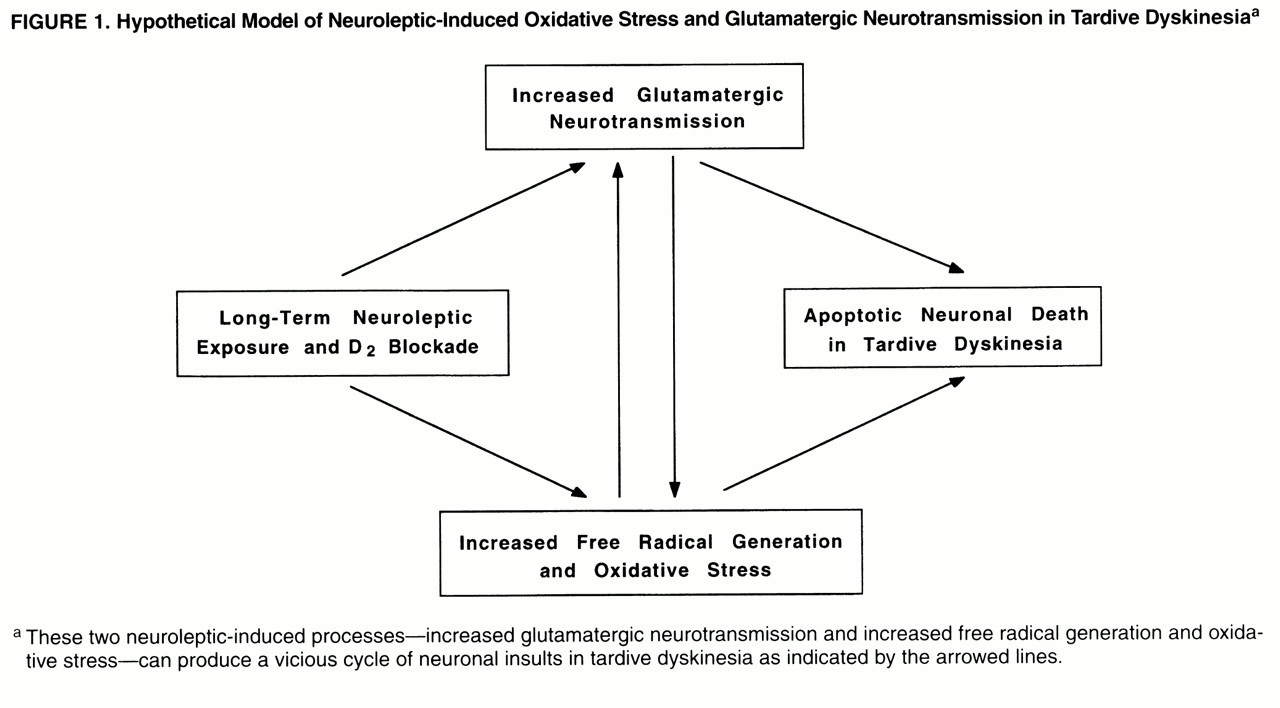
(17). Oxyradicals can damage cellular proteins, membranes, and DNA, depending on their source, causing cell death. An important aspect of the linkage between delayed glutamate-induced degeneration and oxidative stress is that it provides a mechanism whereby persistent low levels of glutamate receptor stimulation can cause cumulative damage to neurons, ultimately leading to degeneration (
figure 1). This form of neurodegeneration exhibits many of the characteristics of “apoptosis.” It provides an important pathological link between moderate levels of excessive glutamate ionotropic receptor stimulation and delayed neuronal degeneration.
Notably, Mitchell et al.
(18) have reported that elimination of striatal dopaminergic afferents leads to striatal neuronal apoptotic death (i.e., program cell death). In addition, elevated oxyradicals can inhibit presynaptic glutamate uptake, inactivate the enzymatic defenses against cellular oxidants
(19), disrupt mitochondrial electron transport
(20,
21), stimulate nitric oxide synthase by the influx of calcium mediated by NMDA receptor, and interact with superoxide to form the highly reactive peroxynitrite radical
(22), which results in an increased generation of redox-active species and extraneuronal excitatory amino acids. These secondary interactions can, thereby, produce a vicious cycle promoting glutamate mediated oxidative damage in the striatum (
figure 1).
The present study was designed to examine the hypothesis that enhanced excitatory amino acid neurotransmission and oxidative damage are present in patients with schizophrenia who received neuroleptic treatment and developed tardive dyskinesia. We also examined the hypothesis that there is a reciprocal relationship between the glutamatergic markers and oxidative stress defense mechanisms (
figure 1). Accordingly, several markers for excitatory neurotransmission (i.e.,
N-acetylaspartylglutamate,
N-acetylaspartate, aspartate, and glutamate) and for oxidative damage (i.e., superoxide dismutase, protein carbonyl content, and lipid hydroperoxides) were measured in the CSF of patients with schizophrenia who had histories of chronic neuroleptic treatment, about half of whom exhibited the symptoms of tardive dyskinesia. Specifically, we hypothesized that concentrations of excitatory neurotransmitters (aspartate, glutamate,
N-acetylaspartate, and
N-acetylaspartylglutamate) are higher in tardive dyskinesia but that superoxide dismutase activity, a defense enzyme for oxidative damage, is attenuated, which results in increased production of oxidized molecules. We also examined the relationships among the glutamatergic markers, oxidative stress markers, and tardive dyskinesia symptoms.
METHOD
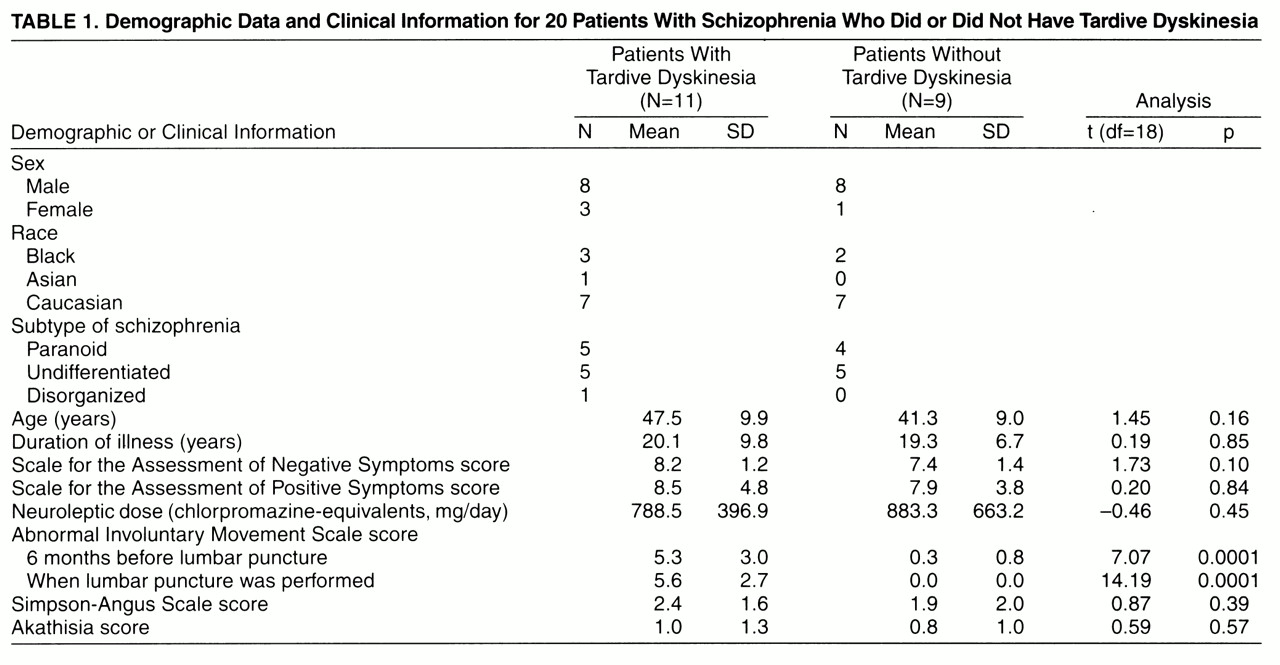
Case control sampling was applied to 20 patients selected from our study group of about 500 outpatients with schizophrenia treated at the Freedom Trail Mental Health Clinic affiliated with Massachusetts General Hospital in Boston. The majority of the patients had chronic schizophrenia; the mean duration of illness was about 20 years (
table 1). After complete description of the study in a protocol approved by our institutional review board, written informed consent was obtained from all subjects. The patients’ tardive dyskinesia status was stratified according to the Schooler criteria
(23) before they enrolled in the study. The severity of tardive dyskinesia was assessed by using the Abnormal Involuntary Movement Scale (AIMS)
(24). The selected subjects were diagnosed by our research psychiatrist (G.T.), and all of them fulfilled DSM-III-R diagnostic criteria for schizophrenia. Exclusion criteria included history of any neurological disorder other than tardive dyskinesia, illicit drug use in the previous 6 months, or any unstable medical condition.
Overall, the patients with and without tardive dyskinesia were similar in their demographics, clinical features, Parkinsonian symptoms, and chlorpromazine-equivalent neuroleptic dose
(25) (
table 1). All of the patients had chronic schizophrenia and had taken neuroleptics for a long time; however, the exact duration of tardive dyskinesia and neuroleptic exposure as well as the life-long doses of neuroleptic could not be ascertained. All of the patients received neuroleptics and had been receiving stable doses during the previous 2 months. In the tardive dyskinesia group, the neuroleptics were fluphenazine (N=5), fluphenazine and thioridazine (N=2), haloperidol (N=2), trifluoperazine (N=1), and thioridazine (N=1). Six tardive dyskinesia patients received benztropine, and one received propranolol. In the non-tardive-dyskinesia group, the neuroleptics were fluphenazine (N=6), fluphenazine and chlorpromazine (N=1), haloperidol (N=1), and haloperidol and trifluoperazine (N=1). Four non-tardive-dyskinesia patients received benztropine, two received trihexyphenidyl, one received clonazepam, and two received lorazepam. Scores on the Scale for the Assessment of Negative Symptoms and the Scale for the Assessment of Positive Symptoms of the two groups were similar (
table 1).
Parkinsonian symptoms were measured by the Simpson-Angus Rating Scale. All of the AIMS and Simpson-Angus Rating Scales were administered by the same investigator. To ascertain the diagnosis, tardive dyskinesia and AIMS scores were assessed once within 6 months before the lumbar puncture and again when lumbar puncture was performed. The AIMS scores at the two assessment periods did not reveal significant changes over time in these patients (
table 1).
Lumbar punctures were performed between 8:00 and 9:00 a.m., following a 12-hour fast. A total of 10 ml of CSF was collected from each patient. Routine analyses were done to confirm that the protein concentration was in the normal range and the cell count was smaller than 5/mm3. CSF specimens were immediately transferred to a –80˚C freezer. All biochemical analyses were performed blind to diagnosis.
To provide a reference group for our study, CSF was obtained from 20 age-matched and sex-matched normal subjects at the Biochemistry Laboratory, Massachusetts General Hospital, to control for psychiatric disorder and neuroleptic treatment. Medical record review was performed to exclude subjects who had histories of neurological disorders, psychiatric disorders, major medical illness, neuroleptic exposure, or illicit drug use. Subjects who had family histories of movement disorders were also excluded.
Amino Acids and Peptide Analysis
Amino acids were measured by
o-phthalaldehyde pre-column derivatization coupled with reverse-phase C-18 column high performance liquid chromatography separation and fluorescent detection
(26). Absolute concentrations of the amino acids were determined by using computer analysis (Maxima 820, Waters, Mass.) of peak height with internal and external standards. The aspartate levels were also reported in another study of this group of patients
(25).
N-Acetylaspartylglutamate and
N-acetylaspartate were measured by anion-exchange high performance liquid chromatography separation and ultraviolet detection at the wavelength of 214 nm
(26). Their concentrations were determined by peak height calculation.
Superoxide Dismutase Analysis
Total superoxide dismutase activity in the CSF was analyzed according to a modification of a spectrophotometric method
(27). The reduction of cytochrome c by superoxide was used to detect and measure superoxide dismutase activity by the change in absorbance at 550 nm. Aliquots of CSF were added to 20 mM of bicarbonate buffer containing 10 M sodium azide, 10 M cytochrome c, 100 M xanthine, and 1 M EDTA. The reduction of acetylated cytochrome c was initiated by the addition of xanthine oxidase.
Lipid Hydroperoxide
The levels of lipid hydroperoxides were measured according to an adaptation of the ferrous oxidation/xylenol orange method described by Puttfarcken et al.
(27). Ten l of CSF was added to 90 l of the reaction mixture (100 M xylene orange (
o-cresolsulfonphthalein-3"3"-bis(methyl-iminodiacetic acid sodium salt), 250 M ferrous chloride, 25 mM H
2SO
4, and 4 mM BHT in 90% methanol and incubated at 30˚C for 30 minutes. The absorbance of the sample was determined at 560 nm against a blank, which consists of all the reagents and 10 l of water. The final concentration of lipid hydroperoxides was calculated using an extinction coefficient of 4.3×10
4 M
–1cm
–1.
Protein Carbonyl Assay
The method for protein carbonyl assay was adapted from Vevine et al.
(28). Briefly, 90 l of CSF was added to 10 l of 10% (weight/volume) streptomycin sulfate in 50 mM of HEPES, pH=7.2, and then centrifuged at 11,000
g for 10 minutes after 15 minutes of incubation. The supernatant was precipitated with an equivalent volume of 20% trichloroacetic acid by centrifuge at 11,000
g for 10 minutes. The protein pellet was redissolved in 50 l of water. Then 6 l of 1 M Tris-HCl, 10 mM of EDTA, and 14 l of 100 mM [
3H]NaBH
4 with a specific activity of 100 mCi/mmol was added to the sample and incubated at 37˚C for 30 minutes. The sample was again precipitated with 1 ml of 10% trichloroacetic acid and centrifuged at 11,000
g for 10 minutes. The pellet was again dissolved in 6 M of guanidine solution. The radioactivity was determined by liquid scintillation counting after incubation at 37˚C for 30 minutes. The data were expressed as picamoles of carbonyl group/g of protein.
Statistics
Because of the small number of patients in the study group, the nonparametric Kruskal-Wallis one-way analysis of variance was computed for each molecule or enzyme activity in CSF between patients with schizophrenia who did or did not have tardive dyskinesia. Bonferroni correction was applied to reduce type I error (
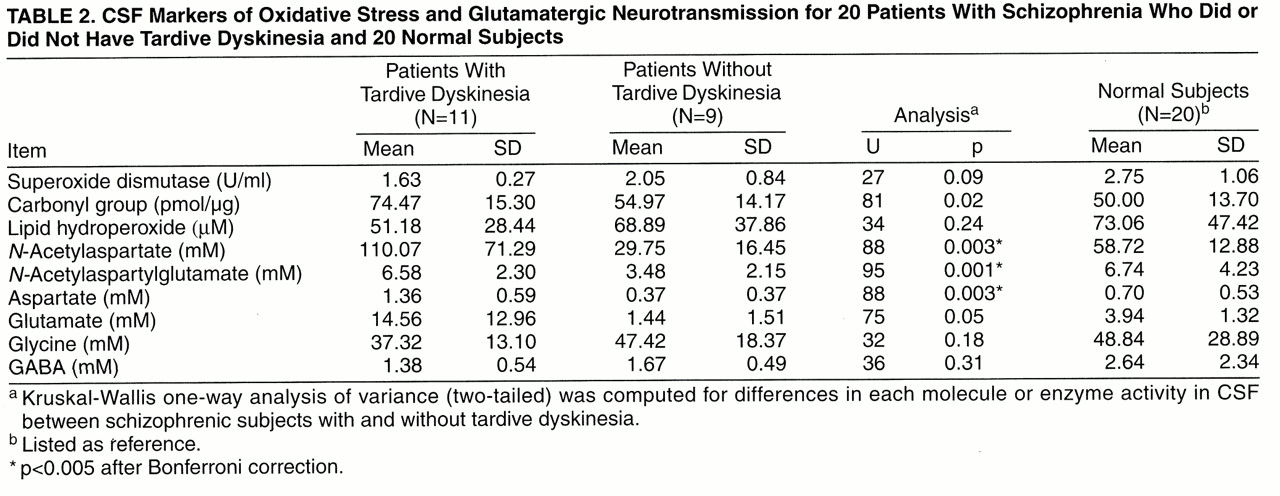
table 2). To test the a priori hypothesis of a vicious cycle promoting glutamate-mediated oxidative damage in tardive dyskinesia, Spearman correlation coefficients were calculated between oxidative stress and glutamatergic markers and also between AIMS scores and CSF markers.
RESULTS
Excitatory Neurotransmitters
After controlling for age and stable dose of neuroleptics, we found that the patients with tardive dyskinesia had higher concentrations of
N-acetylaspartate,
N-acetylaspartylglutamate, and aspartate than the patients without tardive dyskinesia (
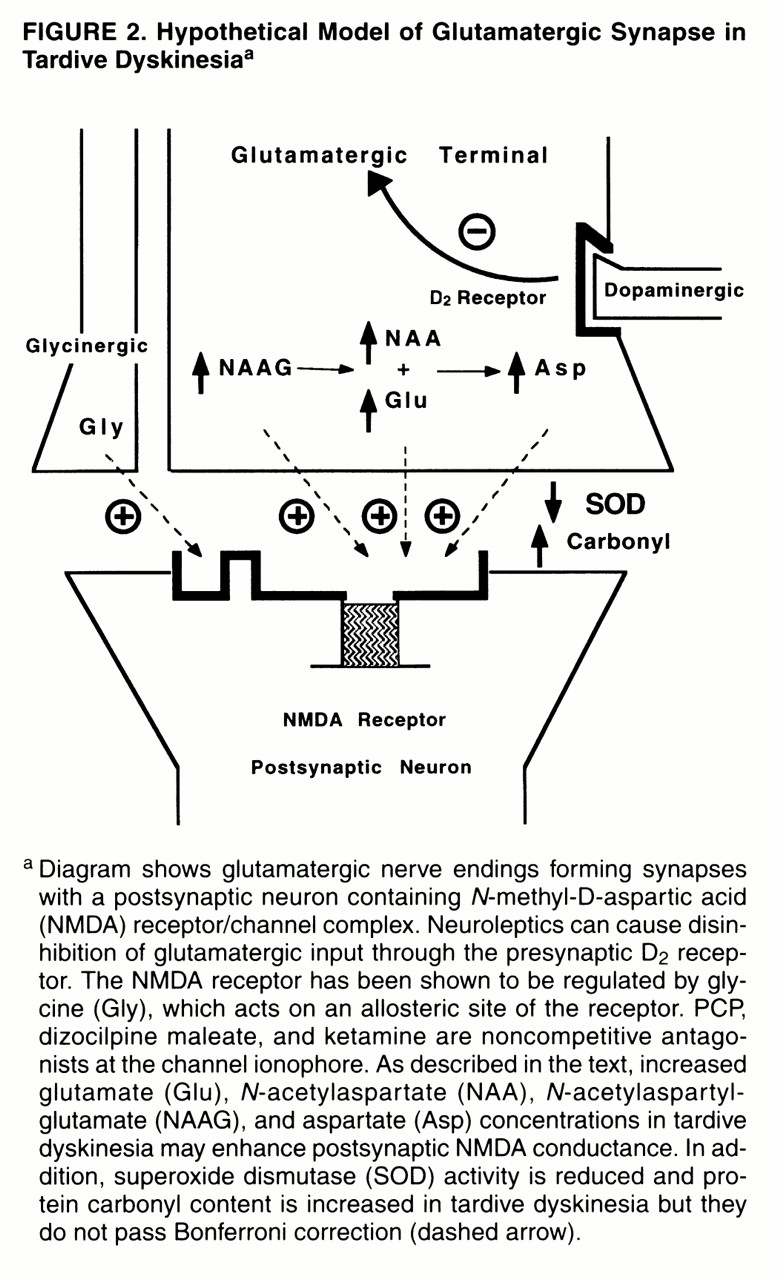
table 2,
figure 2). The elevation in glutamate did not pass Bonferroni correction (U=75, N=20, p=0.05). Levels of glycine and GABA were not significantly different among the three groups (the 20 patients with schizophrenia who did or did not have tardive dyskinesia and the 20 normal subjects from the Biochemistry Laboratory). Benzodiazepine may affect the excitatory neurotransmitters, but the number of subjects receiving benzodiazepine was too small (N=3) to determine its effect.
Oxidative Stress Markers
The CSF of patients with tardive dyskinesia had higher levels of protein carbonyl oxidation products than the CSF of patients without tardive dyskinesia, but this difference did not pass Bonferroni correction (U=81, N=20, p=0.002) (
table 2). Superoxide dismutase activity was lower in the tardive dyskinesia group but did not pass the statistical test (U=47, N=20, p=0.09). However, no difference in the level of lipid hydroperoxides was observed among the patients with schizophrenia who did or did not have tardive dyskinesia and the normal subjects.
Relation Between Oxidative Stress and Excitatory Neurotransmission
In the CSF of schizophrenia patients as a group, superoxide dismutase activity was inversely correlated with aspartate concentration (r=–0.49, N=20, p=0.03); carbonyl group levels positively correlated with aspartate (r=0.45, N=20, p=0.05).
Relation Between Tardive Dyskinesia Presentations and Oxidative Stress and Excitatory Neurotransmission
For all schizophrenic subjects, total scores on the AIMS were positively correlated with N-acetylaspartate (r=0.68, N=20, p=0.001), N-acetylaspartylglutamate (r=0.79, N=20, p=0.0003), aspartate (r=0.63, N=20, p=0.003), glutamate (r=0.44, N=20, p=0.05), and protein carbonyl (r=0.41, N=20, p=0.07) but inversely correlated with superoxide dismutase activity (r=–0.41, N=20, p=0.07). The significant correlations result from the bimodal distribution of AIMS scores in the schizophrenic patients; within the tardive dyskinesia group, AIMS scores did not correlate with any neurochemical markers.
DISCUSSION
Our findings demonstrate higher levels of
N-acetylaspartate,
N-acetylaspartylglutamate, and aspartate in the CSF of patients with schizophrenia who had tardive dyskinesia than in the CSF of patients with schizophrenia who did not have tardive dyskinesia (
table 2). It appears that the elevation in markers for excitatory amino acid neurotransmission relates to the presence of tardive dyskinesia rather than current neuroleptic exposure or underlying psychopathology of schizophrenia because the neuroleptic dose and the status of schizophrenia (subtype as well as positive and negative symptoms) were similar between the patients with and without tardive dyskinesia (
table 1).
Our findings support our a priori hypothesis. However, since all of our subjects had chronic schizophrenia, our findings may not be generalizable to all patients suffering from tardive dyskinesia. In addition, the number of subjects studied was small and statistical power is therefore limited. As a result, there may be type II error. Nevertheless, our findings did reveal the elevation of several excitatory markers (
table 2).
The relatively small number of patients studied did not permit us to examine the effects of the biochemical variables simultaneously, but only in isolation. Therefore, we applied Bonferroni correction to reduce type I error. In addition, the biochemical assays we performed were cross-sectional analyses. The temporal sequence of the onset of tardive dyskinesia and the biochemical changes is not clear, and the case-control design cannot demonstrate a causative relationship.
Previous studies indicated that neuroleptic blockade of presynaptic dopamine D
2 receptors located on corticostriatal glutamatergic afferents enhances the release of glutamate. Ultrastructural study reveals that neuroleptic treatment increases the number of glutamatergic corticostriatal synapses
(29). In fact, Gattaz et al.
(30) reported that the CSF levels of glutamate were higher in patients receiving neuroleptics than those not taking neuroleptics. Consistent with this pathogenic mechanism for elevated striatal excitatory neurotransmission in tardive dyskinesia, clozapine, an atypical neuroleptic rarely causingtardive dyskinesia, increases extracellular aspartate and glutamate in the prefrontal cortex without affecting their levels within thestriatum
(31). The inability of clozapine to increase striatal glutamatergic activity may explain the fact that it is less likely to produce tardive dyskinesia
(14).
Our study also demonstrates a reduction in superoxide dismutase activity and an increase in protein carbonyl concentration in the CSF of tardive dyskinesia patients. Although they did not pass the statistical test, it is important to discuss the findings in the context of the oxidative stress hypothesis of tardive dyskinesia. Superoxide dismutase is an enzyme critical in the detoxification of superoxide, a normal byproduct of oxidative metabolism
(32). Attenuated activity of superoxide dismutase may contribute to the increase of oxidized protein. Chronic treatment of rats with fluphenazine has been reported to cause a decrease in superoxide dismutase and catalase activities in the nervous system
(33). Decreased superoxide dismutase renders neurons more vulnerable to oxyradical injury, consistent with the elevated levels of protein carbonyl groups. Although we did not observe an elevation in lipid hydroperoxides in the CSF of patients with tardive dyskinesia, elevated levels of conjugated dienes and thiobarbituric acid reactive products in the CSF of tardive dyskinesia patients have been reported
(34,
35). The discrepant findings may be due to the different types of patients or analytical methodologies applied.
Superoxide dismutase also protects against neuronal degeneration induced by glutamatergic mechanisms
(36). The inverse correlations between CSF levels of aspartate, protein carbonyl groups, and superoxide dismutase activity in tardive dyskinesia suggest an etiologic relationship between enhanced excitatory amino acid neurotransmission and oxidative damage associated with tardive dyskinesia (
figures 1 and
2). Low peripheral superoxide dismutase activity has been reported in drug-naive patients during a first episode of schizophrenia
(37). In addition to oxidative damage enhanced by neuroleptic-induced hyperglutamatergic neurotransmission, the other plausible model for the development of tardive dyskinesia is that lower activity of superoxide dismutase renders the striatal neurons more vulnerable to excitatory neurotransmission, which is exacerbated by neuroleptics, in a subgroup of patients with schizophrenia.
Our study needs to be replicated with larger groups of patients. The relationships between the glutamatergic and oxidative stress markers can be noncausal. The strong correlations between oxidative stress and glutamatergic neurotransmission suggest that their effects on the occurrence of tardive dyskinesia may be confounded; their strong correlations may preclude separation of their effects statistically even in a large-series study. Also, the findings in CSF may represent only part of the neuronal activity as far as the oxidative damage and enhanced glutamatergic neurotransmission are concerned.
Both preclinical and clinical studies point to degeneration of striatal efferent neurons, especially GABAergic neurons, in tardive dyskinesia. Although inconclusive, one brain imaging study has revealed reduction in the volume of the caudate nuclei in patients with tardive dyskinesia compared with patients without tardive dyskinesia and normal control subjects
(38). In the primate model of experimental tardive dyskinesia, monkeys with neuroleptic-induced dyskinesis exhibit reductions in presynaptic GABAergic markers in the subthalamic nucleus, the medial segment of the globus pallidus, and the rostral part of the substantia nigra
(12). Rodent models of tardive dyskinesia have also revealed significantly lower density of large neurons in the striatum
(39) and decreased glutamic acid decarboxylase activity in the substantia nigra
(11,
40). Finally, Mitchell et al.
(18) demonstrated in the rat apoptotic neuronal death in the striatum as a consequence of a lesion to the nigrostriatal dopaminergic pathway. The elevated levels of CSF
N-acetylaspartate in tardive dyskinesia are consistent with a neuronal degenerative process because CSF
N-acetylaspartate, a marker for neuronal integrity, is elevated in amyotrophic lateral sclerosis
(41) and because tissue
N-acetylaspartate levels decrease in areas involved in active neuronal degeneration in amyotrophic lateral sclerosis, Huntington’s disease, and Alzheimer’s disease (for a review see reference
42)).
The hypothesis of oxidative damage to striatal neurons mediated by neuroleptic enhancement of glutamatergic neurotransmission is supported by reports that vitamin E reverses the symptoms of tardive dyskinesia; the anecdotal reports have been sustained by double-blind, placebo-controlled studies with vitamin E
(43-
45). Notably, patients are more responsive to treatment with vitamin E earlier in the course of their disorder, consistent with the model that the oxidative damage is cumulative over time and would involve functional impairment before frank degeneration. Similarly, in a double-blind, placebo-controlled study of vitamin E treatment for Huntington’s disease, patients who were less symptomatic at the initiation of treatment exhibited the most favorable response
(46). Inasmuch as centrally active free radical scavengers may not only reverse the oxidative damage but also correct an impairment in the inactivation of glutamate, additional studies on the glutamatergic neurotransmission and oxidative stress in tardive dyskinesia as well as efficacy of prevention and treatment with centrally active free radical scavengers on the surrogates of central excitatory neurotransmission and oxidative stress in tardive dyskinesia need to be carried out.