Diagnosis and Natural History
The clinical diagnosis of Prader-Willi syndrome is made on the basis of widely accepted consensus criteria
(7) (appendix 1). This diagnostic scheme relies on a point system that divides signs and symptoms into major and minor criteria. Major criteria are assigned 1 point, with half a point for minor criteria. In addition, there are a number of supportive criteria that do not contribute to the overall total but are helpful in increasing diagnostic certainty. In older children, adolescents, and adults, the diagnosis of Prader-Willi syndrome requires a total of 8 points, with at least 5 from the list of major criteria. In children under the age of 3, diagnosis requires a total of 5 points, with 4 from the major criteria group.
As her background history reveals, Miss A met all major and most of the minor criteria for Prader-Willi syndrome; there were also several other supportive findings. Genetic testing confirmed her clinical diagnosis. While her history and diagnosis are typical and representative of Prader-Willi syndrome, it may be misleading to assume that all cases have such classic presentations. As in Miss A’s case, Prader-Willi syndrome can be diagnosed in the neonatal period, but at other times it may not become evident until much later. The syndrome may first be suspected in adolescents or adults who present with mild mental retardation, obesity, and behavioral difficulties.
One complaint that was not present in Miss A’s history and does not appear among the formal diagnostic criteria but deserves comment is
rectal digging. The problem is common for both children and adults with Prader-Willi syndrome and may present a number of risks, ranging from potential fecal contamination to rectal incompetence and bleeding
(8). The symptom may be related to the skin picking that is nearly ubiquitous in Prader-Willi syndrome. Rectal digging is usually not the most severe problem associated with the disorder but can often be the cause of embarrassment to patients and families. As a result, many times the presence of the symptom will not be mentioned during evaluation and treatment and will not be disclosed unless specifically inquired for.
Physical and behavioral manifestations in Prader-Willi syndrome are highly interrelated. While most physical findings are neither life threatening nor progressive, the sequelae of the syndrome’s behavioral traits are not nearly so benign. In the absence of aggressive intervention, morbid obesity at a young age is common. Food-related behaviors in Prader-Willi syndrome may lead to a host of serious medical consequences, including the range of cardiovascular diseases, diabetes mellitus, sleep disturbances, and respiratory compromise. It is these sequelae that represent the most common causes of premature morbidity and mortality among patients with Prader-Willi syndrome.
Genetics of Prader-Willi Syndrome
Prader-Willi syndrome is a sporadic genetic disorder with an estimated prevalence of 1 in 15,000. It represents the most common diagnosable genetic obesity syndrome
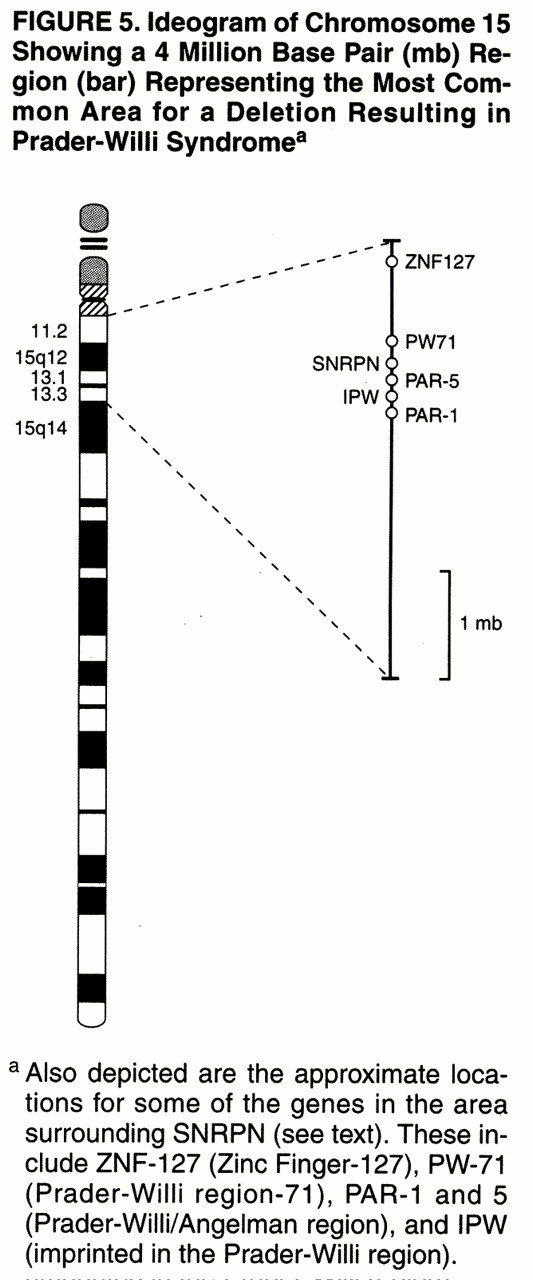
(6). Prader-Willi syndrome results from characteristic abnormalities involving chromosome 15, although the specific gene or genes responsible for the syndrome have not yet been determined conclusively. The search for such a gene or genes has led to the identification of a genetic region of interest and a candidate gene, the small nuclear ribonuclear polypeptide N (SNRPN), which codes for the polypeptide involved in RNA splicing
(9,
10). Other genes in the Prader-Willi syndrome critical region (as those depicted in
figure 5) are likely to be involved. In fact, Prader-Willi syndrome may be considered a contiguous gene syndrome in which a number of nearby loci contribute to its overall clinical phenotype
(11). Genetic research in Prader-Willi syndrome has also uncovered two important genetic mechanisms,
genomic imprinting and
uniparental disomy, that have considerable relevance to the broader understanding of human genetics
(12).
Genomic imprinting and uniparental disomy. A genetic abnormality was documented in Prader-Willi syndrome over two decades after the syndrome was originally identified
(1). High-resolution chromosomal banding allowing for the examination of million base-pair regions of DNA revealed that a substantial portion of patients with Prader-Willi syndrome had large deletions in the proximal long arm of chromosome 15
(13), particularly at bands 11–13 (denoted del 15q11-q13).
A perplexing finding was that an apparently identical deletion of chromosome 15 led to another, and markedly distinct, mental retardation condition, Angelman syndrome
(14,
15). Patients with Angelman syndrome show profound mental retardation, marked language abnormalities, intractable seizures, and physical features impossible to confuse with those of Prader-Willi syndrome. The mystery of seemingly identical genetic lesions leading to two vastly different clinical entities was resolved when it was recognized that in cases of Prader-Willi syndrome, deletions were found only on the chromosome 15 that originated from the father
(16). Conversely, in the case of Angelman syndrome, such deletions occurred only in the maternally derived chromosome. The finding of functional nonequivalence of the parental genomes was unexpected.
This phenomenon, termed
genomic imprinting, describes a situation in which a chromosome retains a “memory” or imprint of its parental origin. The relevance of such a mechanism was emphasized when it was determined that in cases of Prader-Willi syndrome in which no deletion could be found, patients had two normal-appearing maternal copies of chromosome 15 and no paternal contribution, a condition termed
maternal uniparental disomy. Moreover, some patients with Angelman syndrome who did not have identifiable deletions were also found to have paternal uniparental disomy
(17,
18). In short, Prader-Willi syndrome and Angelman syndrome demonstrated for the first time that the difference between maternal and paternal chromosomes in certain specific regions could be highly relevant. The presence of two normal copies of one parental origin can not make up for the absence of the “correct” counterpart chromosome for those particular regions.
One suspected molecular mechanism for imprinting is methylation, a process by which methyl groups bind to cytosine residues, often organized in CpG islands
(12). Allele-specific methylation differences have been documented for a number of imprinted genes, and high degrees of methylation often serve to silence gene expression
(19). Methylation is an epigenetic phenomenon, not resulting in permanent DNA changes, but rather in developmentally timed alterations to the DNA structure that lead to uniparental mono-allelic gene expression. Early in development the preimplantation embryo shows bi-allelic expression and little evidence of methylation. Later in development, tissue-specific patterns of gene expression appear (mono-allelic or bi-allelic expression). For example, the normally imprinted insulin-like growth factor gene shows bi-allelic expression in cells of the choroid plexus
(20). The Prader-Willi syndrome region is differentially methylated depending on the chromosome"s parent of origin: extensively so in the case of maternally derived chromosomes and sparingly in that of paternally derived ones.
The clustering of imprinted genes within specific chromosomal regions that replicate asynchronously has led to the search for DNA regulatory regions, “imprinting centers,” that can extend their effects over large stretches of DNA. In the case of Prader-Willi syndrome and Angelman syndrome, a putative bipartite imprinting center has been identified in the region of the SNRPN promoter
(21). Consistent with this interpretation, mutations in this region result in bi-allelic expression of normally imprinted genes in the Prader-Willi syndrome region. Whether or not the characterization of this region will share features in common with other inactivation control regions such as those found on the X chromosome awaits further study
(22).
Genetic diagnosis and subtyping. The earliest approach to identifying deletions in Prader-Willi syndrome used high-resolution cytogenetic examination, which involves arresting chromosomal division at metaphase, followed by staining and viewing the preparation under a light microscope. Deletions large enough to be seen in this fashion, such as in the case of Miss A, account for 50%–60% of Prader-Willi syndrome cases. Cytogenetic analysis remains useful for identifying these deletions, as well as translocations or ring chromosomes that may also lead to Prader-Willi syndrome.
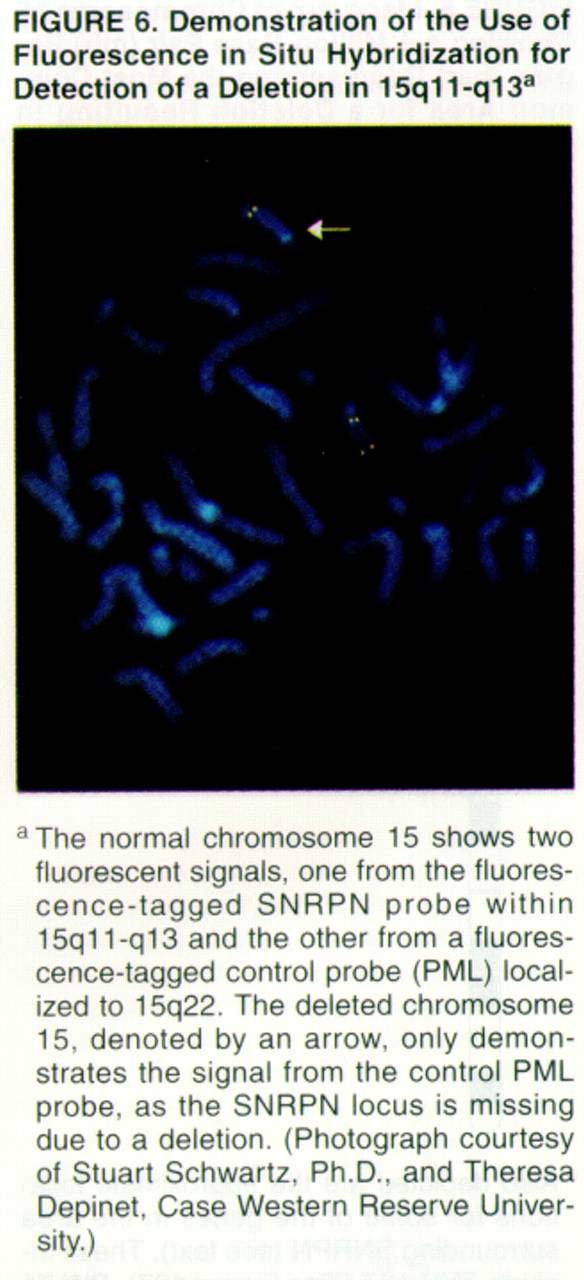
In a significant minority of Prader-Willi syndrome cases, deletions are too small to be visualized in this manner. In such instances, DNA probes are available to search 15q11-q13. Probes with nucleotide sequences complementary to those in the Prader-Willi region, including SNRPN, can be visualized when linked to a fluorescent marker, a technique called fluorescent in situ hybridization (
figure 6).
Despite the much greater resolution afforded by fluorescent in situ hybridization, the approach cannot identify cases of uniparental disomy. Several alternative techniques are able to do so. One older technique relies on restriction fragment length polymorphisms that are able to trace the parent of origin and distinguish between two normal-appearing chromosomes in a suspected uniparental disomy patient. This approach requires blood from the patient and both parents. A more recent alternative technique relies on restriction enzymes that cut DNA differently depending on the methylation status of its regions. The technique requires blood from the patient only and allows for a highly sensitive assay for uniparental disomy, while also being able to identify most deletion cases
(23).
Taken together, these commonly used approaches are effective tools in determining the genetic anomalies associated with Prader-Willi syndrome. However, given the multiplicity of techniques available, clinicians must note that a negative finding on certain examinations does not necessarily preclude either a clinical or a laboratory diagnosis of Prader-Willi syndrome
(23).
The ability to reliably categorize patients on the basis of these techniques raises the question of whether different types of genetic processes lead to distinct clinical phenotypes. While subjects with either the paternal deletion or maternal uniparental disomy share the syndrome"s cardinal features, those with uniparental disomy may show milder symptom expression in both the physical and behavioral arenas
(24-
28).
Neuropsychiatric Aspects of Prader-Willi Syndrome
As reflected in Miss A’s presentation, maladaptive behaviors and psychiatric symptoms in Prader-Willi syndrome are often disproportionately debilitating relative to the cognitive impairment associated with the syndrome. While persons with Prader-Willi syndrome have, on average, mild to moderate levels of cognitive delay, their behavioral and psychiatric dysfunction may overwhelm the patient, the family, and other caregivers or lead to the need for highly restrictive levels of care.
Neurobiological substrates. Core features of Prader-Willi syndrome such as hypogonadism, short stature, and appetite dysregulation have led to suspect hypothalamic dysfunction as a putative neural substrate involved in the disorder. Minor and supportive criteria such as temperature and pain instability and early adrenarche also suggest hypothalamic localization. At present, however, there is no clear evidence for gross anatomical lesions in general, or in the hypothalamus or the pituitary in particular
(29), nor are there any published functional neuroimaging findings.
The notion that the hypothalamus may play a central role in the syndrome has been supported by the single published neuropathological investigation of Prader-Willi syndrome
(30). In postmortem samples, Swaab and colleagues found a significantly lower number of small oxytocin-secreting neurons in the hypothalamic paraventricular nucleus in Prader-Willi syndrome sufferers than in comparison subjects. Arginine-vasopressin neurons, a closely related neuronal population, appeared normal in Prader-Willi syndrome.
Psychiatric phenomenology. While food-related difficulties are the most widely recognized behavioral manifestations of the syndrome, subjects with Prader-Willi syndrome commonly show the types of difficulties that brought Miss A to psychiatric attention: temper tantrums, emotional lability, stubbornness, skin picking, and obsessive-compulsive symptoms. In addition, they may be at increased risk for depressive disorders
(6). All told, these behavioral complaints are sufficiently common to qualify as diagnostic criteria, and they suggest that there may be a distinctive “behavioral phenotype” associated with the disorder.
Dykens and Kasari
(31) compared 43 children with Prader-Willi syndrome aged 4 to 19 years to age- and gender-matched children with Down’s syndrome or nonspecific mental retardation. While all three groups showed stubbornness and temper tantrums, seven behaviors predicted Prader-Willi syndrome group membership with over 90% accuracy: skin picking, obsessions, fatigue, underactivity, impulsivity, speech problems, and talking too much. These same behaviors, along with overeating, also emerged as highly characteristic of a separate sample of Prader-Willi syndrome youngsters relative to subjects with Smith-Magenis syndrome
(32), further showing the distinctiveness of the Prader-Willi syndrome behavioral phenotype.
High rates of non-food-related obsessive-compulsive behaviors were further examined in a study of 91 subjects with Prader-Willi syndrome aged 5 to 47 years
(33). As assessed by the Yale-Brown Obsessive Compulsive Scales, subjects showed an average of three different obsessions and compulsions. Prominent symptoms included hoarding objects, ordering and arranging objects according to certain rules, “just right” phenomena, and repetitive asking or telling. Symptoms were similar in both type and severity to age- and gender-matched patients with obsessive-compulsive disorder (OCD); the subjects with Prader-Willi syndrome were more likely to hoard, and patients with OCD were more likely to show checking compulsions.
Few studies have looked at the relative risk for other types of psychopathology in patients with Prader-Willi syndrome. A recent survey of clinicians familiar with the syndrome estimated the types of psychiatric disorders seen in Prader-Willi syndrome and suggested that as many as 50% of patients may have depressive and anxious symptoms sufficient to warrant a DSM-IV diagnosis
(6). Although such findings have not yet been substantiated by rigorous study, they do suggest that psychiatric disorders present a formidable challenge to patients with Prader-Willi syndrome and their families. A detailed understanding of the common psychiatric and behavioral manifestations of Prader-Willi syndrome may allow families and patients to identify such difficulties and seek treatment early in their course and may help in coping with symptoms that may otherwise be attributed to lazy, stubborn, or manipulative behaviors.
Cognitive profiles. Subjects with Prader-Willi syndrome span the full range of intelligence, from average ability to severe retardation, and the mean IQ of 70 is high relative to those with other genetic mental retardation syndromes. Adaptively, however, even high-functioning individuals rarely function at a level commensurate with their IQs, because of interference from food-related and other behavioral problems. Many individuals with Prader-Willi syndrome show a distinctive, although not necessarily unique, profile of cognitive strengths and weaknesses. Academically, reading/decoding and comprehension may exceed arithmetic skills, although uneven academic performance may not be striking enough to meet learning disability criteria (see reference
34 for review). As in Miss A’s case, some people with Prader-Willi syndrome show relative strengths in spatial-perceptual organization and visual processing tasks; these strengths may relate to the proficiency in jigsaw puzzles noted as a supportive finding in the clinical criteria for Prader-Willi syndrome. By contrast, common weaknesses are noted in sequential processing and short-term memory tasks, including visual, motoric and auditory short-term memory
(35-
37). Not all persons with Prader-Willi syndrome show these profiles, and studies are needed that identify the range and sources of individual differences in cognitive levels and profiles.
Treatment Approach and Prognosis
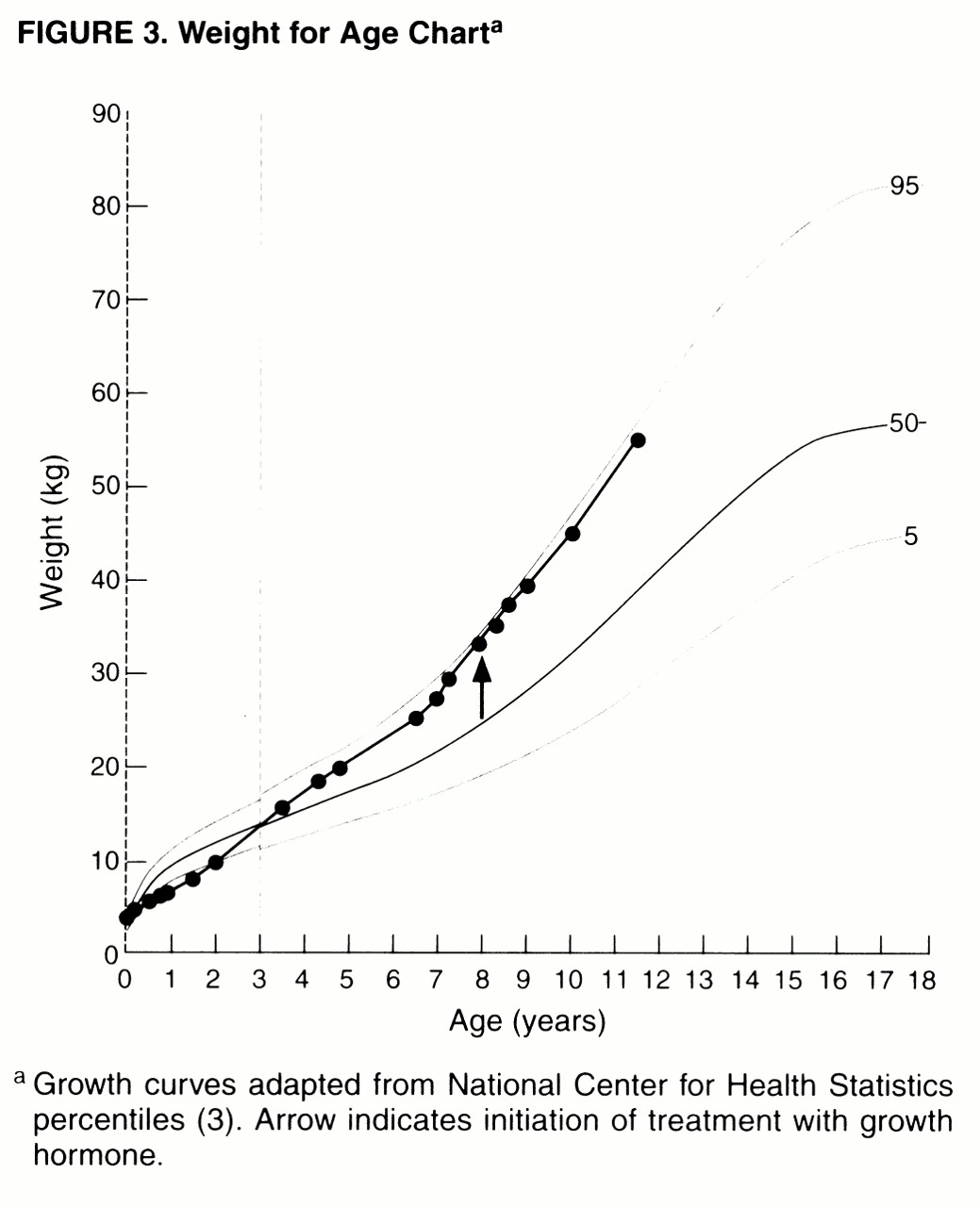
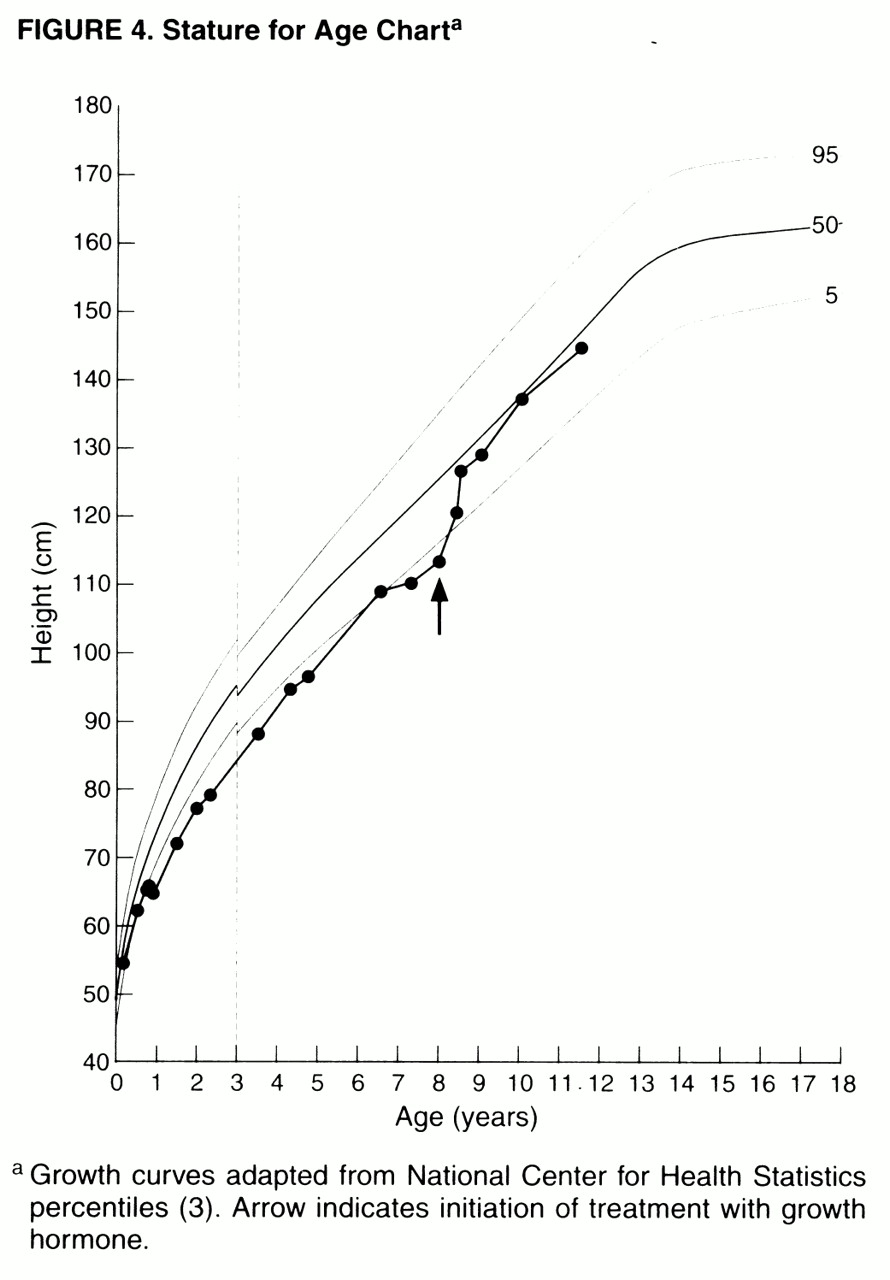
The separation of medical and behavioral aspects in Prader-Willi syndrome is artificial, as exemplified in Miss A’s treatment with GH. GH therapy in children with Prader-Willi syndrome increases muscle tone and enhances growth
(38,
39). Anecdotally, GH is thought to have other significant benefits, such as allowing certain patients to moderately increase daily caloric intake without incurring substantial weight gain, thus mitigating some of the difficulties associated with maintaining highly restrictive diets. Through GH’s combined effects on muscle mass and tone, and in increasing height and decreasing central obesity, it can have a substantial effect in normalizing body habitus (
figure 2). As in Miss A’s case, these combined results can have an enormous impact on the psychological well-being of Prader-Willi syndrome patients and their families, as well as decreasing their risks for medical morbidity.
In contrast to the clear evidence supporting the use of GH, there is a paucity of studies of anorectic agents in the management of Prader-Willi syndrome, and anecdotal reports have been uniformly discouraging. When phentermine, fenfluramine, and dexfenfluramine were available, there was little to suggest their efficacy in Prader-Willi syndrome, especially over time. Now that the severe cardiovascular consequences of these agents are known
(40), their use in Prader-Willi syndrome is not supportable.
From the standpoint of non-food-related behavioral problems in Prader-Willi syndrome, there are few guidelines regarding psychopharmacology. The relatively high rate of anxious and depressive presentations among Prader-Willi syndrome patients has led to the frequent use of SSRIs. Marked improvements in the repetitive, aggressive, and affective symptoms of Prader-Willi syndrome through use of these medications have been reported in small open studies
(41-
43), although controlled trials are lacking. One frequent observation has been that doses expected for body weight appear to be poorly tolerated in Prader-Willi syndrome. As in Miss A’s case, a number of patients have shown exacerbations of aggressive, repetitive, and compulsive behaviors during initiation or upward dose titration of medication. Despite the generally positive experience with SSRIs for a variety of complaints common to Prader-Willi syndrome, there is unfortunately little to suggest that these medications have an impact on eating patterns or weight gain.
The role of antiepileptic agents, opiate antagonists, and benzodiazepines, as sometimes used in the treatment of impulsive behavior in patients with mental retardation, is not well documented in Prader-Willi syndrome
(44). Clinically, neuroleptics are widely used and are an option for treatment-refractory patients, but they should be reserved as second-line agents because of their potential for unwanted side effects, including further weight gain.
Even with the clinical success seen with SSRIs and GH, the mainstay of management for behavioral difficulties—both food- and non-food-related—remains behavior modification. Programs such as token economies or star systems may be useful
(44). Efforts to restrict food intake by careful dietary planning, close supervision, and limiting food access must extend out of the home and into school, work, and community settings. Hoarding food and stealing money to buy food are extremely common complaints in Prader-Willi syndrome, and vigilance is required even when patients are otherwise extremely well behaved. Exercise programs are also important and must be appropriate to the cognitive level and physical skills of the patient.
The management of the overall range of behavioral problems seen in Prader-Willi syndrome may be quite taxing of families, teachers, and patients. Out-of-home placement often becomes necessary over time
(6,
44). Certain “dedicated” group homes specialize in the treatment of individuals with Prader-Willi syndrome, and in cases in which obesity, other behaviors, or both are out of control, such highly structured and specialized programs may be critical. Leaving the family home for a group setting may be viewed as a transition that fulfills developmental needs not dissimilar from those of other young adults. Nonetheless, out-of-home placement is at times construed by families and patients as a sign of failure. These feelings may be exacerbated by the fact that the potentially life-threatening problems associated with Prader-Willi syndrome typically arise from behaviors usually thought of as willful and controllable. Patients with Prader-Willi syndrome may feel that they are not trying hard enough to control their urges or their temper, and their families may feel that they have failed because obesity and tantrums remain a problem. The clinician can help to place these issues into a developmental context, to reinforce the idea that blame need not be assigned, and to remain engaged with patients and their families over time.
Prader-Willi Syndrome as a Heuristic Model
Efforts to understand the epigenetic mechanisms underlying Prader-Willi syndrome have led in unexpected directions. One of the most intriguing observations is that paternally imprinted genes differentially contribute to the development of hypothalamic and septal regions of the brain, while maternally imprinted genes contribute to the development of the neocortex and striatum
(45). While these observations fit nicely with the clinical phenotypes associated with Prader-Willi syndrome and Angelman syndrome, respectively, the basis of this differential contribution remains enigmatic. Remarkably, from an evolutionary point of view, genomic imprinting is a relatively recent phenomenon—seen only in mammals and not in other metazoa, including other vertebrate species.
Since vertebrate development is highly conserved and imprinting is a mechanism found mostly in mammals, it is thought by some that imprinting mechanisms are unlikely to be “necessary” for the development of the well-known structures contained within the vertebrate CNS
(46). On the other hand, it is clear that among mammalian species this mechanism for adjusting gene dosage has profoundly influenced aspects of brain development and growth. On the basis of morphometric analyses, some investigators have suggested that genomic imprinting may be one of the mechanisms responsible for the nonlinear expansion of neocortical and striatal structures in primate evolution
(47).
One of the more provocative proposals to account for why the mammalian genome is ornamented with as many as ~3×10
7 methyl groups concerns the competition between parental genomes, particularly when the paternity of the offspring is in doubt
(48,
49). Although this proposal is speculative, it is consistent with the observation that imprinted genes do not appear to be strictly conserved across mammalian species (see reference
46 for a review). Indeed, there is some evidence that patterns of genomic imprinting may reflect both species-specific patterns of social and reproductive behavior and differences in brain morphometry
(45). The finding in Prader-Willi syndrome of reduced oxytocinergic neurons in the paraventricular nucleus
(30) may be consistent with this proposal, given the role of oxytocinergic pathways in the initiation of maternal and other affiliative behaviors (see reference
50 for a review). These apparent alterations in oxytocin levels may also play a role in the obsessive-compulsive symptoms, and in the separation distress seen in Miss A and other patients with Prader-Willi syndrome
(51,
52). This set of observations has also led us to suspect that genomic imprinting may play a role in OCD unrelated to Prader-Willi syndrome, and a search of the Prader-Willi syndrome region of chromosome 15 for genes that might contribute to OCD is underway. Finally, novel therapies directed toward hypothalamic dysfunction, and possibly involving congeners of oxytocin, may prove to be of value in this syndrome.