As already noted, not every human postmortem study has shown a high number of dopamine uptake sites, perhaps because of use of different radioligands to evaluate the dopamine transporter (
4) or because of a coexisting loss of dopamine neurons in severely cocaine-dependent individuals (
5). Differing results with two radioligands both specific for the dopamine transporter have also occurred in animal studies (
7–
12), suggesting that the affinity of dopamine transporter binding sites may be changing. In the present experiments we evaluated the biochemical nature of the abnormalities in dopamine transporter binding by using the cocaine analog [
3H]WIN 35428 in 15 cocaine-using subjects and 15 matched comparison subjects to confirm that the first group had a higher number of binding sites and to determine whether any change had occurred in the dopamine transporter’s affinity for dopamine and cocaine, as well as for WIN 35428. We also assayed binding to the vesicular monoamine transporter type 2 (VMAT2) by using the radioligand (+)-[
3H]dihydrotetrabenazine (DTBZ), an independent marker of dopamine terminal density that is not directly regulated by monoaminergic drug treatments (
13). We further examined differences between groups in [
3H]WIN 35428 binding for possible relationships to clinical and demographic features, which have not been previously examined. Our initial hypotheses were that the number of [
3H]WIN 35428 binding sites (B
max) would be higher in the cocaine-using subjects, without differences in dopamine transporter affinity, and that the difference between groups would be superimposed on a lower number of dopamine terminals in the cocaine users. We further hypothesized that subjects with more severe cocaine dependence would demonstrate greater abnormalities in [
3H]WIN 35428 binding.
METHOD
Subjects
Fifteen pairs of cocaine users and matched comparison subjects were chosen at random from a larger group comprising 34 pairs whose [
3H]WIN 35428 binding had been previously examined autoradiographically (
3). This was done to conserve tissue and laboratory resources after a statistical power calculation indicated that a subset of subjects would provide sufficient power to examine the issues of interest. The subset of randomly chosen paired subjects were found to be representative: among the larger group, cocaine users had 46.3% greater [
3H]WIN 35428 binding autoradiographically in the caudate than the matched comparison subjects, 40.1% greater binding in the putamen, and 44.6% greater binding in the accumbens. In the subset used in the present experiments, the cocaine users displayed 46.6% greater binding in the caudate than the matched comparison subjects, 44.5% greater binding in the putamen, and 49.6% greater binding in the accumbens.
The methods used to obtain brain samples have been previously described (
1,
3) and will be briefly reviewed. Postmortem brain specimens were obtained at autopsy as authorized by the Wayne County (Michigan) Medical Examiner’s Office, freshly dissected, quickly frozen on dry ice, and stored at –70°C until sectioned. The subjects had died suddenly in accidents, by assault, or of cardiovascular causes. The comparison subjects and cocaine users were pair-matched for postmortem interval, age, sex, and race, as well as for socioeconomic status to control for any nonspecific factors associated with impoverished nutrition and living conditions. The comparison subjects met the following criteria: 1) suitable method of death (rapid, not the result of a chronic condition); 2) appropriate age, sex, and race to balance the cocaine-using subjects; and 3) absence of DSM-IV diagnoses other than ethanol abuse or nicotine use. The cocaine users were without evidence of chronic medical illness and exhibited a range of typical psychiatric symptoms and psychoactive substance use disorders. In addition to cocaine, some subjects were also diagnosed as ethanol abusers (seven cocaine users, five comparison subjects), nicotine users (five cocaine users, four comparison subjects), or opioid users (four cocaine users only).
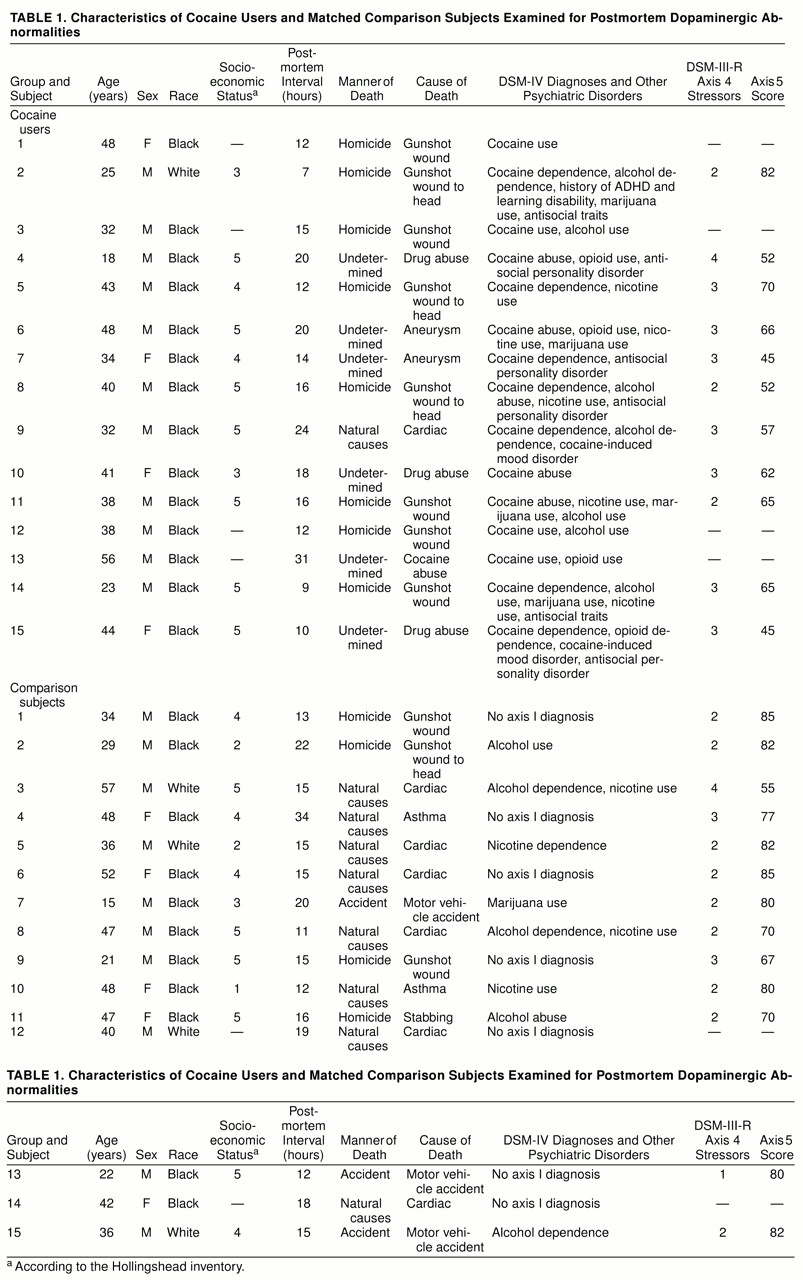
Table 1 lists the cause of death for each subject, as well as demographic and diagnostic information.
Clinical Assessment
A research psychiatrist and clinical social worker interviewed at least one knowledgeable informant for each subject, most often a first-degree relative. Other types of informants included neighbors, friends, fellow workers, police officers, medical examiners, physicians, mental health personnel, and newspaper reporters. Family members who participated in the study provided informed consent under guidelines approved by the Institutional Review Board of the University of Michigan. The interviews focused on ascertaining whether the subject met the DSM-IV criteria for any psychoactive substance use disorder, alcoholism, antisocial personality, mood disorder, anxiety disorder, or psychotic disorder. A Hollingshead rating of socioeconomic status was derived for every subject. Based on the available evidence, DSM-IV psychiatric diagnoses were assigned at a consensus conference. When further questions remained, efforts were renewed to discover the specific information needed, and the diagnostic assignment was postponed. The subjects’ cocaine dependence was quantified in three ways. Two scales were used to rate overall severity of use, both acutely and chronically. The subject’s use was coded as mild, moderate, or severe and assigned 1, 2, or 3 points on the basis of review of all available evidence. In addition, axis 5 scores were assigned by using the standard directions, to provide an estimate of overall dysfunction. Both psychiatrists involved (K.Y.L., G.W.D.) were experienced in the use of these scales and were knowledgeable regarding the issues involved in quantifying the effects of drug use (
14). Diagnoses and scores were assigned before the assays were performed.
Toxicology
Urine or serum from each subject was assayed qualitatively for the presence of cocaine, opioids, antidepressants, antipsychotics, and anxiolytics by means of a variety of methods, including radioimmunoassay, high-performance liquid chromatography, and gas chromatography/mass spectroscopy. Ethanol levels were measured by dichromate microdiffusion/gas chromatography methods.
Binding Assays
The methods used to assay [
3H]WIN 35428 binding (also referred to as [
3H]CFT) have been previously described and will be briefly reviewed (
1). Because the earlier autoradiographic assay had shown that the degrees of dopamine transporter regulation by cocaine were similar in all regions of the striatum, tissue from either the caudate or putamen was initially homogenized in buffer solution (50 mM Tris, 120 mM NaCl, pH 7.4). The homogenate was centrifuged at 2,000 rpm for 5 minutes, the precipitant was discarded, and then two high-speed centrifugations (17,000 rpm for 20 minutes) were performed, resulting in a washed pellet. After incubation at a final tissue concentration of 5.0 mg/ml (conditions to be described), binding reactions were terminated by rapid filtration. The filters were washed twice, and their radioactivity was counted. Nonspecific binding was defined by the addition of 30 µM (–)cocaine. Nonspecific binding with [
3H]WIN 35428 averaged 15%–20% as a fraction of total binding in striatum. Protein concentrations were determined by using a commercial assay kit (BioRad, Claremont, Calif.).
Saturation experiments, using 16 concentrations of WIN 35428 from 10–12 to 10–5 were performed at the equilibrium conditions determined previously (2°C for 60 minutes) with 3 nM of [3H]WIN 35428 (specific activity, 80 Ci/mmol; New England Nuclear/Dupont, Boston). Saturation experiments were performed by homologous displacement because of the cost of [3H]WIN 35428 and the high nonspecific binding encountered at higher [3H]WIN 35428 concentrations. [3H]WIN 35428 competition experiments again used 3-nM concentrations of [3H]WIN 35428 and eight concentrations of the competitors and were performed at 2°C for 60 minutes. WIN 35428 was supplied by Dr. Ivy Carroll (Research Triangle Institute, Research Triangle Park, N.C.). (–)Cocaine and dopamine were purchased from Sigma Chemicals (St. Louis).
The autoradiographic assay methods for (+)-[
3H]dihydrotetrabenazine (DTBZ) have been previously described (
13). Briefly, 16-µm-thick sections were incubated at 2°C in Tris buffer (50 mM, 120 mM NaCl, pH 7.4) for 1.5 hours. An initial saturation study indicated a dissociation constant (K
d) of 1.9 nM (SD=0.1) in human striatum. On the basis of this result, a single saturating concentration of [
3H]DTBZ (10 nM) was used to assess binding in the striatum, by means of two total and one nonspecific sections. Nonspecific binding was defined with reserpine, 10 mM. After incubation, the slides were washed and then apposed to Kodak SB-5 film for 5–7 days. Amersham tritium microscale standards (Amersham, Arlington Heights, Ill.) were co-exposed for each cassette. Optical densities were determined by using an image analysis system (Microcomputer Imaging Devices, Ottawa, Ont., Canada). Optical densities were evaluated by comparison to the Amersham standards and are expressed in nanocuries per milligram.
Data Analysis
Computer analysis of the binding data was performed with Prism Software version 2.0 (GraphPAD Software, San Diego), by using iterative curve-fitting techniques. A coefficient of determination was calculated for each set of data as an indicator of how well the data fit the function, similar to a correlational coefficient. Saturation and competition data were fit to one- or two-component models and evaluated for goodness of fit by F test. Further data analysis used SPSS version 6.1.3 (SPSS, Chicago). [3H]WIN 35428 and [3H]DTBZ binding were confirmed initially to be normally distributed (Kolmogorov-Smirnov distances were calculated, p>0.10 in all cases). [3H]WIN 35428 Bmax and Kd, the cocaine concentration causing 50% inhibition of binding (IC50), dopamine IC50, and [3H]DTBZ binding levels were compared in the cocaine users and comparison subjects by group t tests (after an initial evaluation revealed that the factors on which subject pairing was based—age, postmortem interval, sex, and race—did not affect the neurochemical variables). [3H]WIN 35428 binding and [3H]DTBZ levels were further examined for correlative relationships. In addition, [3H]WIN 35428 binding and [3H]DTBZ were examined for relationships with alcohol diagnosis (both alone and with cocaine diagnosis) by two-way analysis of variance (ANOVA) followed by univariate tests. Other clinical data, including age, postmortem interval, sex, race, and other psychiatric diagnosis (psychosis, mood disorder, opioid use), were examined for effects on [3H]WIN 35428 binding and [3H]DTBZ by using ANOVA or correlational analysis, as appropriate. Initially, to confirm that the cocaine users and comparison subjects had been well matched, age, sex, race, socioeconomic status, and postmortem interval were also compared by using two-tailed t tests.
RESULTS
The cocaine users and comparison subjects were not significantly different in overall demographic characteristics. Their mean ages were 37.3 years (SD=10.3) and 38.3 (SD=12.3), respectively. Most of the subjects in both groups were male (73% of the cocaine users and 67% of the comparison subjects) and African American (93% of the cocaine users and 73% of the comparison subjects; t=2.17, df=28, p=0.15). The mean ratings of socioeconomic status were similar: 4.4 (SD=0.8) for the cocaine users and 3.8 (SD=1.4) for the comparison subjects (t=2.12, df=22, p=0.16). Neither were the postmortem intervals significantly different in the two groups (cocaine users: mean=15.7 hours, SD=6.2; comparison subjects: mean=16.8 hours, SD=5.7). However, both axis 4 and axis 5 mean scores differed between groups. For axis 4 the mean scores were 2.8 (SD=0.6) for the cocaine users and 2.2 (SD=0.7) for the comparison subjects (t=4.54, df=22, p<0.05). For axis 5 the mean scores were 60.1 (SD=11.2) and 76.5 (SD=2.4), respectively (t=16.34, df=22, p<0.01). Individual values for the preceding variables are shown in table 1.
The [
3H]WIN 35428 saturation experiments resulted in a series of curves from 30 individuals that were all curve fit with a coefficient of determination of >0.97 (R
2, the percentage of variance accounted for). In all but one of these experiments a one-site model was preferred.
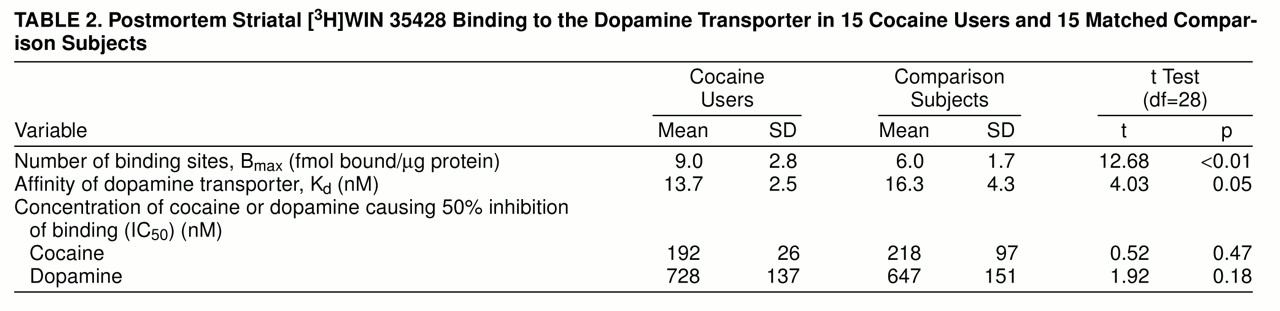
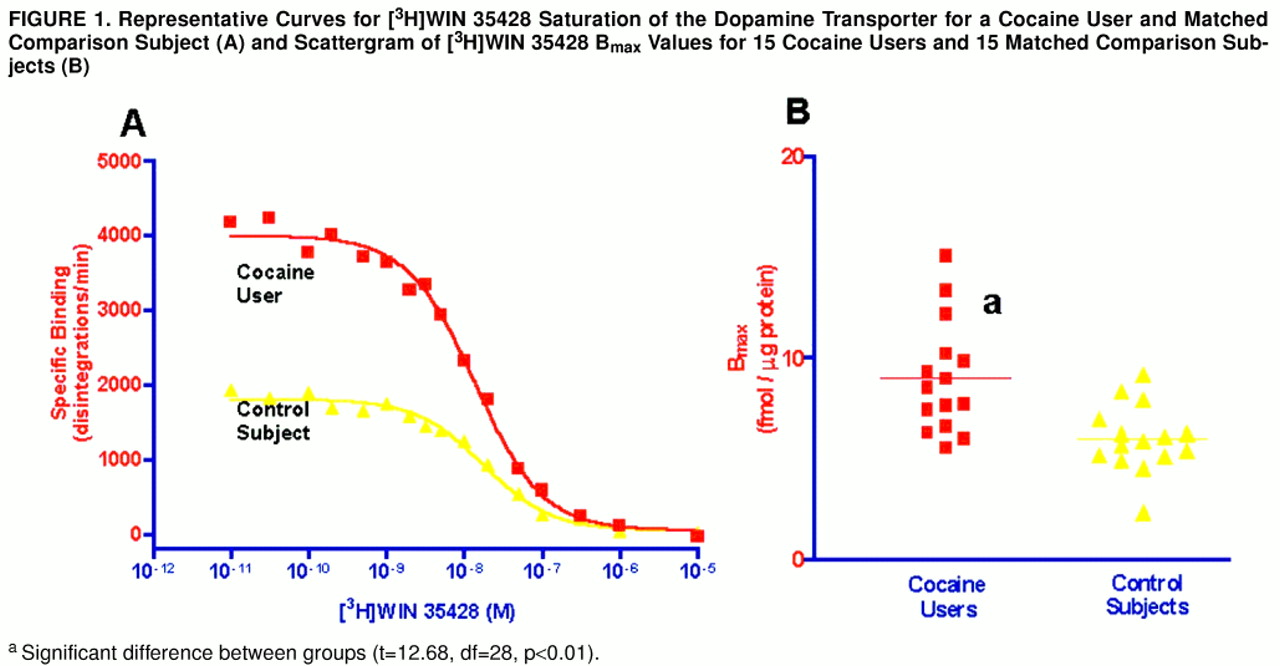
Figure 1A displays the curves fit to the data derived from a matched cocaine user and comparison subject. Similarly, the series of competition curves for the effects of cocaine and dopamine on [
3H]WIN 35428 binding were all best fit to one-site models. There were significantly more striatal [
3H]WIN 35428 binding sites in the cocaine users than in the comparison subjects (
table 2); figure 1B is a scattergram of results. Dopamine transporter affinity for [
3H]WIN 35428 appeared slightly increased in the cocaine users than in the comparison subjects, but the difference was not statistically significant (p=0.054). The potencies of cocaine and dopamine in displacing [
3H]WIN 35428 binding from the dopamine transporter were similar in the two groups (table 2). Age and postmortem interval were also examined for correlation with [
3H]WIN 35428 B
max in all subjects and in the cocaine users alone. Neither age (r=0.01, N=30, n.s.) nor postmortem interval (r=–0.03, N=30, n.s.) was related to [
3H]WIN 35428 B
max.
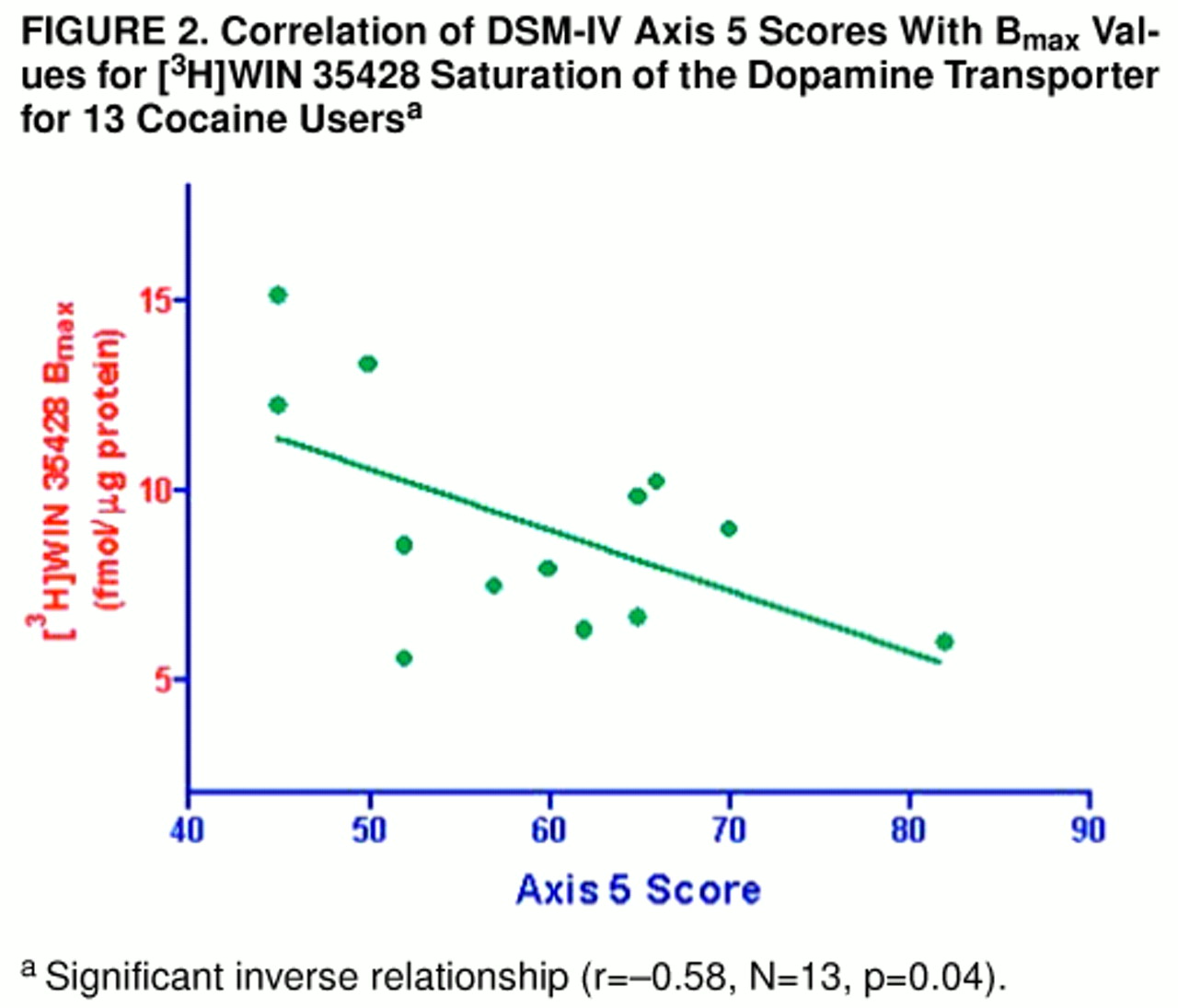
Among the cocaine users (as shown in
figure 2), axis 5 score was significantly and inversely correlated with [
3H]WIN 35428 B
max (r=–0.58, N=13, p=0.04). Severity of chronic use (r=0.82, N=14, p<0.05) and of acute use (r=0.50, N=15, p=0.07) also appeared related. Ten subjects were retrospectively rated as moderate cocaine users chronically, three were rated as having severe use, while two could not be rated with sufficient confidence. The three subjects rated as having the most severe use displayed [
3H]WIN 35428 B
max values that were first, second, and fourth highest among all subjects.
Across all subjects, those with alcohol-related diagnoses displayed a mean [3H]WIN 35428 Bmax of 6.4 fmol bound/µg protein (SD=2.1), whereas alcohol nonusers had a mean value of 8.0 (SD=2.8) (t=2.62, df=28, p=0.12). A two-way ANOVA examining the interaction of cocaine and ethanol exposure confirmed that there was a significant cocaine effect (F=13.46, df=1, 29, p<0.01) and also showed a nearly significant ethanol effect (F=3.67, df=1, 29, p=0.07). However, there was clear evidence against the existence of a significant two-way interaction between cocaine and ethanol (F=0.01, df=1, 29, p=0.93). Between the ethanol abusers and nonusers there was no difference in [3H]WIN 35428 Kd, cocaine IC50, or dopamine IC50. [3H]WIN 35428 Bmax appeared higher, but not significantly, in users of both opioids and cocaine (mean=10.4 fmol bound/µg protein, SD=3.3) compared to users of cocaine only (mean=8.5, SD=2.1) (t=1.43, df=13, p=0.25). Overall, nicotine users had Bmax values (mean=7.5, SD=3.1) similar to those of nonusers (mean=7.4, SD=1.9) (t=0.53, df=28, p=0.48), and among the cocaine users only there was also no difference between nicotine users (mean=8.2, SD=2.1) and nonusers (mean=9.4, SD=3.1) (t=0.01, df=13, p=0.92).
[3H]DTBZ binding was significantly less in the cocaine users (mean=330 nCi/mg, SD=42) than in the comparison subjects (mean=374 nCi/mg, SD=68) (t=4.40, df=28, p<0.05). A correlational analysis did not show a significant relationship between dopamine transporter and vesicular monoamine transporter binding levels (r=–0.34, N=28, p=0.08). Neither age or postmortem interval had an effect on [3H]DTBZ binding level. There was not a significant relationship between [3H]DTBZ binding and the cocaine use severity indices (axis 5: r=0.48, N=13, p=0.11; chronic severity: r=–0.26, N=14, p=0.42; acute severity r=–0.22, N=15, p=0.48). VMAT2 binding in the ethanol-abusing comparison subjects appeared somewhat lower (mean=340 nCi/mg, SD=48) than in the nonusers (mean=388, SD=72), but the finding was inconsistent (t=1.47, df=13, p=0.25). The relationship appeared reversed among the cocaine users: the mean VMAT2 binding was 349 nCi/mg (SD=32) for those alcohol-related diagnoses but 315 (SD=44) for the ethanol nonusers (t=2.66, df=13, p=0.13). Neither opioid use nor nicotine use was associated with abnormal VMAT2 binding.
DISCUSSION
The current results, showing 50% more [
3H]WIN 35428 binding sites in cocaine users than in comparison subjects by means of homogenate techniques, closely replicate the earlier autoradiographic results for these same subjects, which indicated 46% more binding sites (
3). Our results provide new biochemical information suggesting that the greater binding is not caused by a significant difference in the dopamine transporter’s affinity for [
3H]WIN 35428, cocaine, or dopamine. The present results clearly confirm that there are more dopamine transporter binding sites on dopaminergic neurons, despite apparently fewer total dopamine terminals, as measured by [
3H]DTBZ. Our results further replicate the findings of an earlier, smaller autoradiographic study we performed with samples from a different set of subjects who died in a different region of the United States (
1).
Similar results have been reported by Staley et al. (
2) for human striatum. In contrast to Staley et al., we found evidence for only a single type of [
3H]WIN 35428 binding site on the dopamine transporter, with a single affinity. In our experience, lower sodium concentrations in assay buffer (as used by Staley et al.) induce a large number of lower-affinity [
3H]WIN 35428 binding sites (
15). Despite intensive experimentation, it remains unclear why different laboratories obtain differing models for dopamine transporter binding. In the experiments showing two sites, the reported affinity constants for the two sites are fairly close (
16,
17) and would be expected to be difficult to detect on theoretical grounds. In any case, both the present study and the Staley et al. study (
2) confirm that it is high-affinity dopamine transporter sites that are up-regulated in chronic cocaine users. Mash et al. (
18) have also recently found that an increase in striatal dopamine uptake occurs in human cocaine users, paralleling [
3H]WIN 35428 binding site abnormalities.
In contrast, Wilson et al. (
5) did not find significant greater [
3H]WIN 35428 binding among 12 cocaine users postmortem than in matched comparison subjects. This finding is puzzling because these same investigators reported greater [
3H]WIN 35428 binding in rats treated with cocaine (
7). One possible explanation is that the human subjects were suffering from a superimposed, more marked loss of dopaminergic neurons, as evidenced by 22% less VMAT2 binding, contrasting with the 12% lower binding found in the present subjects. Greater severity might reflect the process through which the samples were collected in the Wilson et al. study. Five forensic medical centers across North America each selected tissue samples from two or three subjects that were most confidently diagnosed retrospectively as cocaine users. In this way, the selection of more behaviorally disturbed individuals may have occurred. Despite the fact that there were 22% fewer [
3H]DTBZ binding sites in the postmortem samples examined by Wilson et al. than in comparison subjects, [
3H]WIN 35428 binding was 6% higher. This indicates that the number of [
3H]WIN 35428 binding sites per dopamine terminal was also high in these subjects.
The low [
3H]mazindol binding reported by Hurd and Herkenham (
4) suggests that [
3H]mazindol binding sites are not up-regulated by cocaine exposure and therefore reflect the overall loss of dopaminergic terminals. Hitri et al. (
19) also have reported that binding of [
3H]GBR12935 is low in the frontal cortex of human cocaine users. Although potent inhibitors of dopamine uptake, both mazindol and GBR12935 are structurally dissimilar from cocaine and WIN 35428. Other experiments (
7–
12,
20) have consistently shown that various dopamine-transporter-selective compounds are not similarly regulated by cocaine exposure. Understanding how some, but not all, binding sites on the dopamine transporter can be increased will require the development of a complex molecular model that takes into account these distinctive stoichiometric changes.
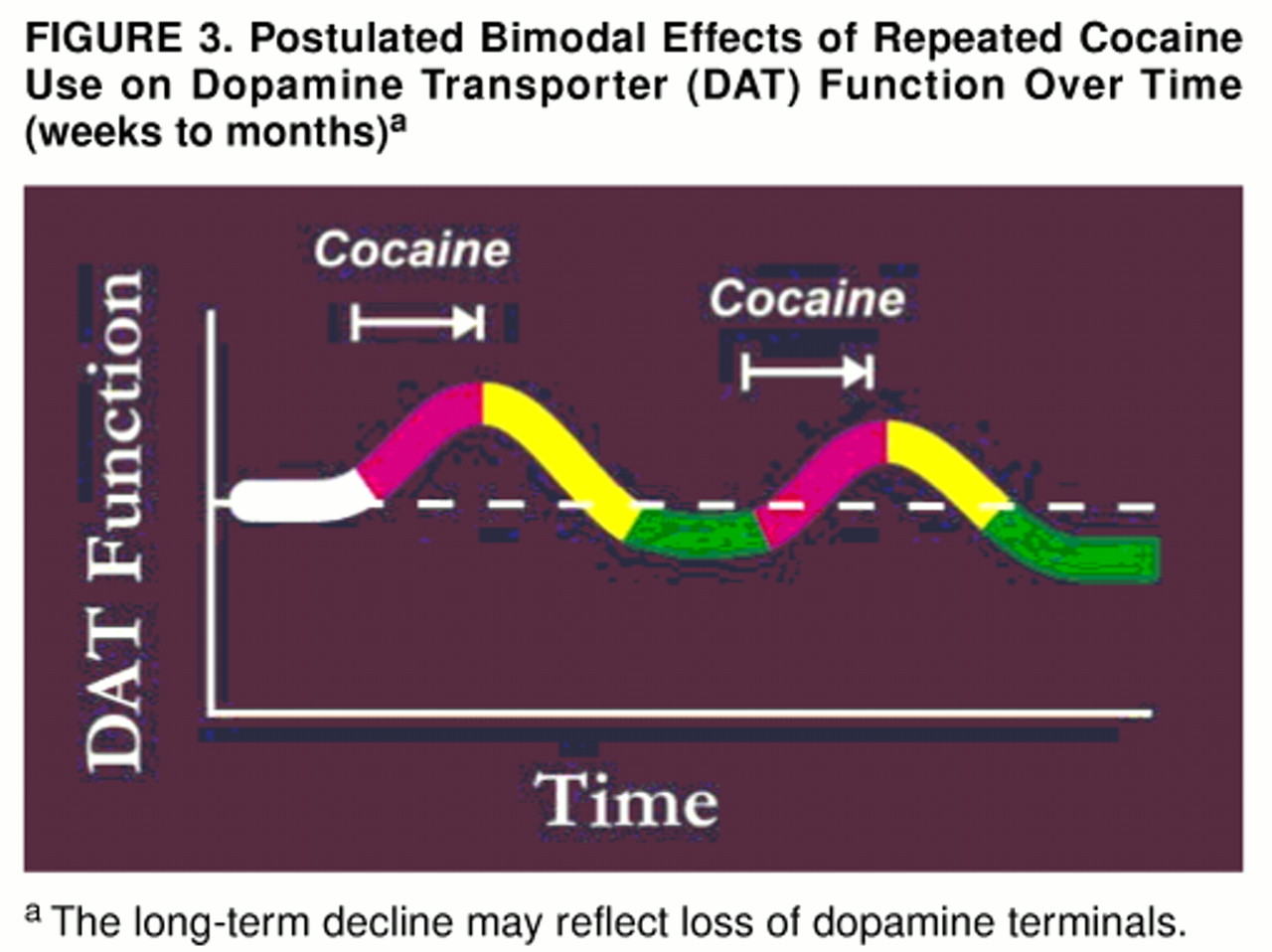
As noted already, the large number of [
3H]WIN 35428 binding sites found in the present group of subjects occurred despite a lower number of [
3H]DTBZ binding sites. (The postulated temporal relationship between an increase in the former and a decrease in the latter is diagrammed in
figure 3.) As noted earlier, the loss of [
3H]DTBZ binding sites may reflect a decrease in total number of dopaminergic axons. Animal studies (
21–
23) have suggested that cocaine is not neurotoxic, but our results and those of Wilson et al. (
5) and Volkow et al. (
24) (who found positron emission tomography evidence for decreased dopamine transporter binding in subjects free of cocaine for many weeks) together suggest that neuronal damage may occur in humans who abuse cocaine. Such a postulated axonal loss may be directly related to cocaine exposure or to other concomitant factors, including ethanol, opioids, other drugs of abuse, and nutritional changes. Our examination of these other factors, in fact, did suggest that ethanol dependence is associated with a lower number of [
3H]WIN 35428 binding sites, in both the cocaine users and comparison subjects. This finding is similar to the results found in the larger subject group, where binding in the putamen was significantly less among alcoholic than nonalcoholic comparison subjects (
3). Surprisingly, however, VMAT2 binding was not comparably lower in ethanol abusers, a finding that is difficult to reconcile. One possible explanation is that alcoholism was underdiagnosed among the cocaine users. However, efforts were made to avoid this mistake, and a similar dopamine transporter/VMAT2 disjunction existed among non-cocaine-using subjects as well. Although it is unlikely that VMAT2 is regulated by monoaminergic-active drugs (
13,
23), determining whether or not low [
3H]DTBZ binding truly reflects a loss of dopamine axons will require further confirmation by means of quantitative morphometric techniques.
There was a suggestion of possibly greater dopamine transporter [
3H]WIN 35428 binding among opioid users, but our numbers were too small and opioid use was too entangled with severity of cocaine use to be clearly distinguished. Previous experiments in rats have shown that chronic morphine treatment markedly alters dopaminergic cell morphology, diminishing neuronal size and rearranging internal architecture (
25). Such changes could possibly affect [
3H]WIN 35428 binding sites, and the possibility deserves further attention. In distinction to opioid use, we found no evidence that nicotine use affects [
3H]WIN 35428 or [
3H]DTBZ binding.
To our knowledge, the present results are the first to show a relationship among the severity of cocaine intake, behavioral dysfunction, and abnormalities in striatal [
3H]WIN 35428 binding to the dopamine transporter. The causal relationship among these variables is unclear but of obvious interest. A simple explanation is that increased cocaine intake induces greater [
3H]WIN 35428 binding site up-regulation. A dose-response relationship has not been previously described in the animal literature but is plausible. An alternative and clinically provocative interpretation is that the individuals who most readily up-regulated [
3H]WIN 35428 binding were those prone to more intense withdrawal effects, bingeing, and addictive behavior. Whether there are marked individual differences in sensitivity to dopamine transporter regulation is unknown but could conceivably exist. Recent evidence (
26) suggests that genetic differences in the human serotonin transporter promoter region alter the adaptive response to chronic ethanol exposure. These considerations further imply that individual differences in serotonin transporter response to chronic antidepressant inhibition might also occur and contribute to therapeutic failures, loss of response, and nonresponse to higher doses.
Not as clear a relationship was found between low [3H]DTBZ binding and the behavioral measures. However, a loss of dopaminergic terminals, if that is the true significance of low [3H]DTBZ binding, could certainly contribute to chronic alterations in reward experience and drug craving. A more extensive exploration of VMAT2 abnormalities in chronic cocaine and ethanol users is now being undertaken.
The present experiments indicate that dopamine uptake is highly sensitive to chronic blockade by cocaine and that the adaptive mechanisms induced in the dopamine transporter are complex. These changes may contribute to the alterations in subjective experience and behavior characteristic of chronic cocaine abusers. Our data further suggest that damage to dopaminergic neurons, which are critical to normal reward processes, may result from chronic cocaine abuse.