Haloperidol is one of the most frequently prescribed neuroleptic agents. The serum elimination of haloperidol in humans slows with time after dosing, and near-terminal elimination half-life may be measured in days rather than hours
(4,
5). A comparable effect is evident in experimental animals. A single dose of haloperidol resulted in signs of central dopaminergic blockade for at least 20 days after administration
(6–
8). Campbell and co-workers
(9) found a half-life of 7.6 days for the disappearance of antidopaminergic effects of a single injection of haloperidol in rats. Similarly, Cohen and colleagues
(6,
7) directly measured behavioral effects and brain haloperidol concentrations in rats after single doses of haloperidol. They found a terminal elimination half-life of the drug in the brain of 6.6 days
(7) and 16.7 days
(6). These results may explain the prolonged clinical effects of neuroleptic drugs even after short-term treatment.
It is not known whether the extended effects of neuroleptic drugs in humans are due to the continued presence of drug in tissue or to long-lasting drug-induced physiologic changes. The aim of the present study was to directly examine haloperidol concentrations in human brain tissue in relation to drug-free time.
METHOD
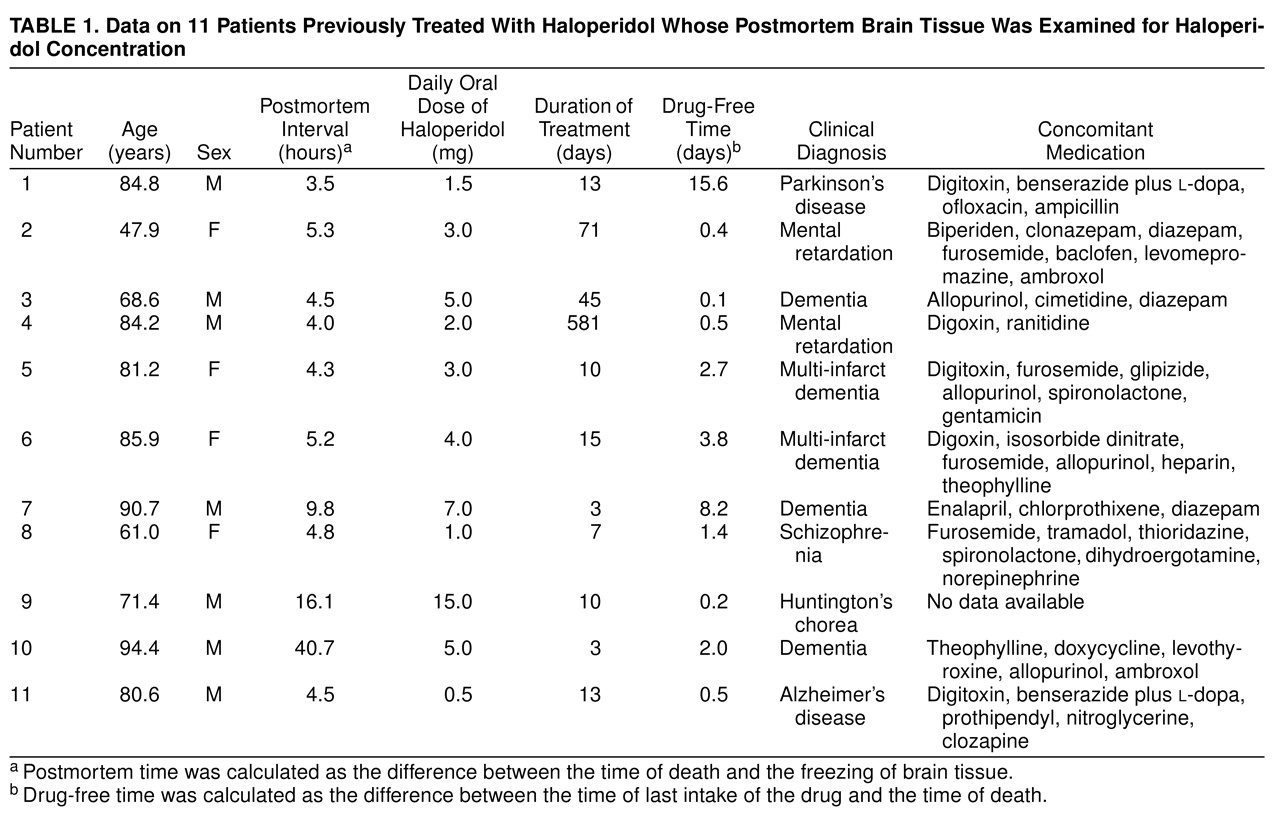
Brain tissue was taken at autopsy from 11 subjects who had been treated with oral doses of haloperidol. The brains were collected between 1989 and 1994. Case notes were examined in detail to establish the duration and dosage of haloperidol as well as the drug-free time before death. The drug-free time was calculated as the difference between the time of last intake of the drug and the time of death. The postmortem time was calculated as the difference between the time of death and the freezing of brain tissue. The histories of these patients, with prescribed drug dosages, duration of treatment, drug-free time before death, and concomitant medication are shown in
Table 1. Tissue samples were taken from several brain regions from either side of the brain (temporal cortex, cingulate gyrus, caudate nucleus, dentate nucleus, corpus callosum); it was not possible to investigate tissue samples of all these regions from every brain. Brain samples were also collected from patients who had never received haloperidol. Postmortem handling of the autopsy material was similar in all cases and was performed according to a standardized procedure
(10). The samples were placed in a freezer at –80°C until analysis. The duration of storage at low temperature was between 2.2 and 7.1 years.
Extraction and Quantitation of Haloperidol
Techniques of extraction, chromatography, and detection were modified from those in a previous publication
(11). Brain samples were cleaned of visible blood and weighed, and 0.4–0.9 g were homogenized with the use of a Sonifier (Branson-250, Danbury, Conn.) in 1 M NaHCO
3 together with 60 ng of the internal standard, bromperidol. The alkalinized homogenate was extracted with hexane/isoamyl alcohol (98.5%/1.5% by volume) by vigorous rotary action. The layers were separated by centrifugation, and the organic phase was aspirated. The butyrophenones were back-extracted into dilute acid and, after alkalinization, again extracted into the hexane/isoamyl alcohol mixture. The resulting organic phase was evaporated under a gentle stream of nitrogen, and the tubes were sealed and stored in darkness at 4°C until high-performance liquid chromatography analysis, which was performed within 2 days. All samples were reconstituted in 150 µl of the mobile phase, and 100 µl were injected into the high-performance liquid chromatography system.
The high-performance liquid chromatography system consisted of a Constametric 4100 solvent delivery system (LDC Analytical, Gelnhausen, Germany), a manual injection valve with a 100-µl loop (Rheodyne Sokati, Rohnert Park, Calif.), an Astec-Pak 5-µM spherical C8 cartridge column (4.6 × 15 cm; Astec, Whippany, N.J.), and a Spectromonitor 4100-wavelength ultraviolet detector (LDC Analytical). The column was operated at 21°C, and ultraviolet detection was performed at 247 nm. The mobile phase consisted of acetonitrile/water (50%/50% by volume; high-performance liquid chromatography grade) titrated to pH 6.5 with 85% orthophosphoric acid. The mobile phase was pumped at a rate of 2.0 ml/min. Chromatograms were recorded on an IBM-compatible personal computer with LDC Lctalk Software. Haloperidol and bromperidol had stable and reproducible retention times that allowed identification and quantitation without interference of endogenous or other psychotropic substances. The concentrations of haloperidol were calculated by using manually determined peak height ratios of haloperidol to the internal standard, bromperidol. Haloperidol content was expressed relative to the wet weight of brain tissue (nanograms per gram).
The extraction recovery was assessed by adding a standard amount of haloperidol to brain tissue of patients without a known history of haloperidol medication. Absolute recovery of the extraction of haloperidol from brain tissue was 93.5% (SD=5.4%). Within- and between-day coefficients of variation in human brain were below 10%. The detection limit was about 4 ng/g. The peak height of haloperidol showed a linear relation to concentrations over a range of at least 4–3000 ng/g. Calibration curves were consistently linear and passed through the origin.
Bromperidol was donated by Janssen Research Products (Beerse, Belgium). Haloperidol was purchased from Sigma Pharmaceuticals (Deisenhofen, Germany). All other chemicals were purchased from Merck and Co. (Darmstadt, Germany) and were of the purest grade available.
Statistical Analysis
Data were analyzed in an exploratory manner. Since there was only a single measurement per individual in various brain regions and insufficient data to describe the pharmacokinetic profile in each individual patient, a population pharmacokinetic analysis
(12) was performed with the computer software program NONMEM
(13). The aim of this analysis was to estimate the average half-life of haloperidol in the brain tissue of the group of patients investigated. Several assumptions had to be made. Steady-state conditions in brain tissue were assumed when the duration of treatment was 7 or more days; otherwise, multiple dosing was modeled. The residual variability was fixed to the estimated coefficient of variation of the analytical method, that is, 10%. Individual Bayes post hoc estimates
(14) were calculated for the volumes of distribution. For the mean subject in the study group, a dose of 4 mg of haloperidol and steady-state conditions were used. Subroutines ADVAN2 and TRANS1 of the package NONMEM IV, version 2.1, were used. The pharmacokinetic model was C=D·1000/V·e
(–k·t), where C=concentration (ng/g), D=dose of drug (mg), V=volume of distribution (liters), k=elimination constant (1/hour), and t=drug-free time after last dose (days).
The model describes the concentration time curves in a one-compartment model after bolus injection. The estimation of an absorption constant was not attempted because of the low number of data points. The analysis used mean concentrations of haloperidol in brain tissue.
Correlations between mean haloperidol concentrations and independent variables were assessed by the Spearman test. Mean values are reported.
RESULTS
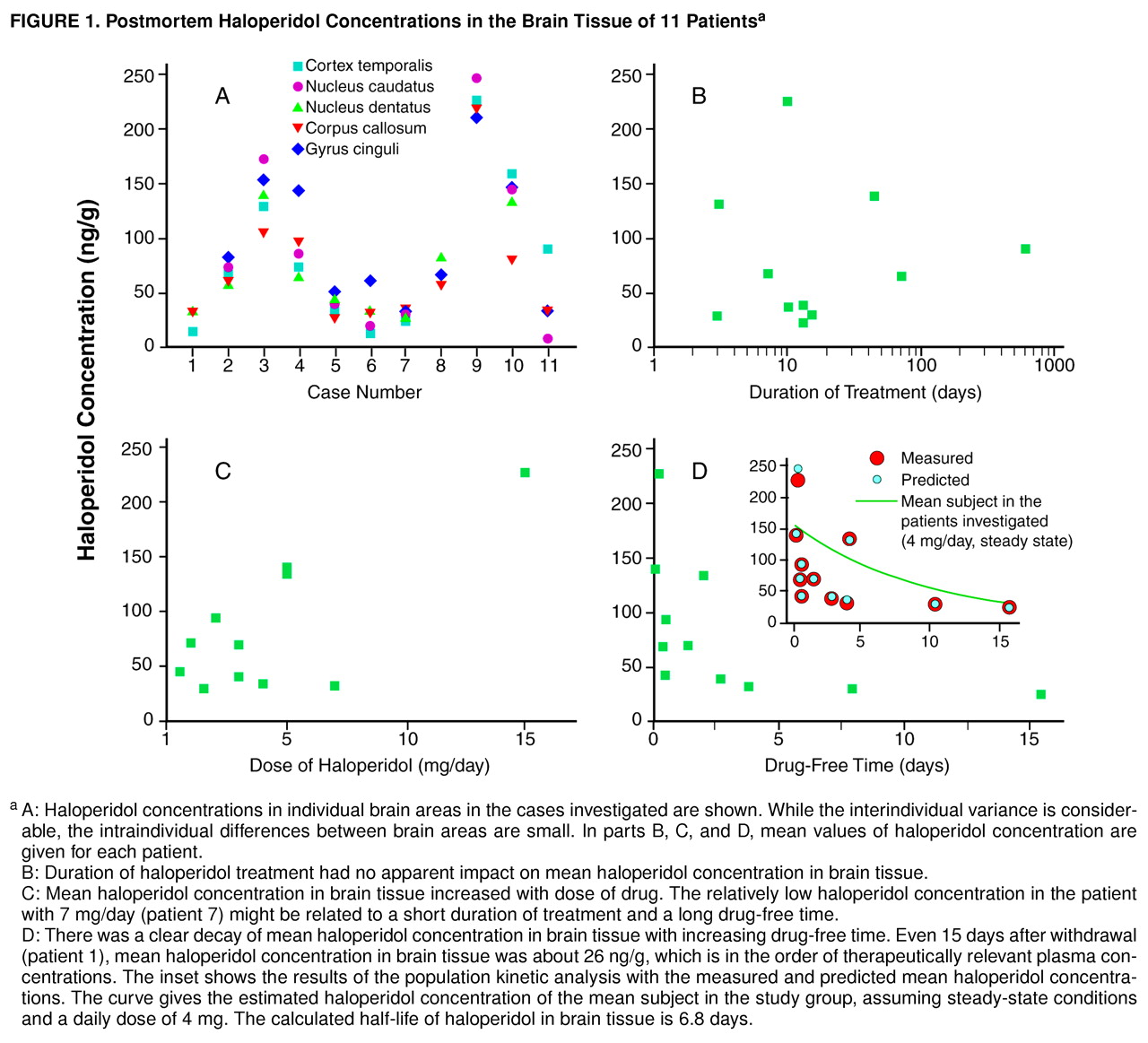
The haloperidol concentration was below the detection limit in the three brain tissue samples of patients never treated with haloperidol. Haloperidol was detected in the brain tissue of all patients previously treated with the drug, with concentrations ranging from 8.9 ng/g (caudate nucleus, patient 11) to 226.7 ng/g (temporal cortex, patient 9). Haloperidol concentrations appeared to be homogeneously distributed across different brain areas within a single individual (
Figure 1, A). To investigate interindividual differences, mean values of haloperidol concentrations were calculated for every patient. In contrast to the intraindividual differences between several brain regions, the interindividual differences between patients were considerable. The highest mean haloperidol concentration was found in a patient on a relatively high dose of haloperidol that had been given until death (patient 9). Low concentrations in brain tissue were found when haloperidol had been withdrawn for more than a week (patients 1 and 7).
Figure 1 shows mean haloperidol concentrations in brain tissue in relation to duration of treatment, dose of the drug, and drug-free time before death. There was no apparent relation between duration of treatment and mean haloperidol concentration. Even after treatment for only 3 days, considerable brain concentrations were measured (
Figure 1, B). Higher doses of haloperidol seemed to be related to higher concentrations in brain tissue (
Figure 1, C). The brain concentration decreased within days after drug withdrawal (
Figure 1, D). In the population kinetic analysis with NONMEM (inset in
Figure 1, D) a brain elimination half-life of 6.8 days was calculated for the mean subject (k=0.00427 1/hour, V=266.1). The asymptotic standard deviation, as an estimate of the precision of the calculated elimination constant, was obtained from the variance-covariance matrix by using the NONMEM program. The asymptotic standard deviation of the elimination constant was 0.00164, resulting in a range of brain elimination half-lives from 4.9 to 11.0 days. Postmortem time was not significantly related to mean haloperidol concentration (r=0.35, N=11, p=0.29). Furthermore, there was no significant correlation between storage time of brain tissue at –80°C and mean haloperidol concentration (r=–0.37, N=11, p=0.26), which is consistent with a high chemical stability of haloperidol during prolonged storage
(15).
DISCUSSION
We measured haloperidol concentrations in human postmortem brain tissue. Because of its high selectivity and sensitivity, a high-performance liquid chromatography method was used for quantification of haloperidol. High-performance liquid chromatography methods are well-suited to situations like this, where several different drugs have to be reliably separated from each other
(11,
16). Korpi et al.
(17) previously quantified haloperidol concentrations in postmortem human brain tissue. That study used a smaller sample of cases and was undertaken to understand the conversion of haloperidol to the reduced alcoholic metabolite in human brain tissue. These authors did not further investigate the factors that might have an influence on haloperidol concentrations. The present study was undertaken to understand the elimination pharmacokinetics of haloperidol in human brain tissue.
We had the opportunity to investigate two patients in our series who had prolonged drug-free time. Much of the data depends on these individuals. The main finding of our investigation is that the brain elimination half-life of haloperidol is about 1 week. Therefore, it is likely that the slow disappearance of neuroleptic effects after the discontinuation of therapy is related to a slow elimination of the drug from the brain. However, the persistence of physiologic effects after complete clearance of the drug may also occur. The estimated elimination half-life of haloperidol from human brain tissue is in close agreement with values derived from the measurement of brain haloperidol concentrations
(7,
18) and antidopaminergic effects
(7,
9) in experimental animals. The elimination half-life appears to be longer in brain tissue compared with serum values, even when one is examining terminal elimination half-lives of haloperidol
(4,
5,
19).
The time course of removal of neuroleptic drugs from human brain tissue has been indirectly estimated through D
2 dopamine receptor occupancy in human patients by using postmortem D
2 receptor measurements, in vivo positron emission tomography (PET), and in vivo single photon emission computed tomography. In postmortem studies, high dissociation constant (K
d) values for the D
2 receptor reflect residual neuroleptic drug at the receptor. A decline of K
d values to control levels within 2 weeks after withdrawal of neuroleptic drugs has been reported for postmortem human brain putamen
(20). Baron and co-workers
(21,
22), using PET, reported on normal receptor availability within 5–15 days after withdrawal of oral neuroleptics. In the PET examination of a single patient, Farde and colleagues
(23) found that after withdrawal of oral haloperidol for up to 54 hours, there was a rapid fall in serum drug levels without a significant reduction in D
2 receptor occupancy. Withdrawal of depot preparations of fluphenazine decanoate
(24) and haloperidol decanoate
(25) resulted in high D
2 dopamine receptor occupancy for several months.
The concentrations of haloperidol reported here are of the same order of magnitude as previously reported values in human
(17) and rat
(6,
17,
26,
27) brain tissue. The concentrations reported by Korpi et al.
(17) for the human brain were in the range between 64 and 3947 ng/g. The highest concentrations reported were 10 times higher than the concentrations found in the present study. However, the patients in that study received higher doses of haloperidol (up to 100 mg/day) and may have had very short drug-free times. When the brain concentrations measured in the present study are compared with clinically effective plasma haloperidol levels reported in the literature
(28,
29), the brain-to-blood concentration ratio in humans is about 10–30, even after short-term treatment. Similar concentration ratios were found by Korpi et al.
(17) in two subjects. These results are paralleled in animal experiments. A brain-to-blood concentration ratio of 22 for haloperidol was found in rats both after a single dose
(27) and after subchronic administration
(26).
Haloperidol appeared to be homogeneously distributed across the brain areas investigated. It is unlikely that a homogeneous distribution happened secondarily during the postmortem time. The postmortem interval was less than 6 hours in all but three cases. Previous data
(17) suggest that an unequal equilibrium concentration of butyrophenones in certain body areas is retained after death. Korpi and co-workers
(17) found no consistent differences in haloperidol concentration across three regions of two postmortem human brains. Similar results have been obtained with the rat brain, where no relevant differences in the concentration of haloperidol between the striatum, limbic system, and cerebellum after a single intraperitoneal injection were found
(18). On the other hand, in experimental animals, a preferential accumulation of butyrophenones in brain areas with a large dopamine concentration has previously been suggested
(17,
30,
31).
It has been hypothesized that the therapeutic delay of antidepressant and neuroleptic drugs might be related to a slow uptake of the drug into human brain tissue
(32). A slow uptake has been found for amantadine in postmortem human brain tissue
(33) and for fluoxetine in vivo
(34). We did not find a slow uptake for haloperidol in the present study. Even after 3 days of treatment with haloperidol, the concentration in brain tissue was comparable to that for long-term treatment. This indicates that haloperidol rapidly accumulates in brain tissue. Similar results were found in experimental animals. As stated above, high brain concentrations of haloperidol were found even after a single administration of the drug in rats
(6,
17,
27), while subchronic treatment did not result in a further increase of the blood-to-brain concentration ratio
(17,
26).
During long-term therapy with neuroleptic drugs, about 30% of patients develop tardive dyskinesia, which remains as a chronic disease after withdrawal of the drug in a substantial portion of the patients. The cause of tardive dyskinesia has been associated with neurotoxic properties of neuroleptic drugs. Recently, similarities between the metabolism of haloperidol and the dopaminergic pro-neurotoxin 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) have been identified
(35). Haloperidol is similar in structure to MPTP, which is metabolized to the neurotoxic 1-methyl-4-phenylpyridinium species (MPP
+). Haloperidol itself is also metabolized to a pyridinium metabolite, 4-(4-chlorophenyl)-1-4-fluorophenyl-4-oxobutylpyridinium (HPP
+)
(36,
37). HPP
+ and MPP
+ have neurotoxic effects on dopaminergic and serotonergic neurons
(37–
39). The neurotoxic pyridinium metabolite is also found in human brain
(40,
41) and may contribute to slowly appearing and irreversible extrapyramidal side effects that occur after long-term treatment. The high concentrations and the long half-life of haloperidol in human brain tissue found in the present study suggest that neurotoxic metabolites may be present in brain tissue for several weeks, even after short-term treatment.
In conclusion, we directly measured concentrations of haloperidol under therapeutic conditions in human brain tissue. Concentrations in brain tissue are 10–30 times higher than the optimum serum concentrations in the treatment of schizophrenia. The estimated elimination half-life of the drug in brain tissue is 6.8 days. After two half-lives (about 2 weeks) there is still a considerable amount of haloperidol in brain tissue. The brain concentration at this time is in the order of therapeutically relevant plasma concentrations. The results reported here may have implications for clinical treatment decisions and the design of clinical research protocols. Patients exposed to haloperidol cannot be considered to be free of residual effects of the drug for a number of weeks after withdrawal, even after acute treatment and even when the drug concentration is below the detection level in the blood.