Schizophrenia is a disabling mental disorder that affects 1% of the population worldwide
(1) . The symptoms of schizophrenia cause pervasive impairments in many domains of functioning and are costly in terms of the personal suffering of patients and families and the burden on the health care system. The symptoms and impairments of schizophrenia likely reflect neurodevelopmental deviations that lead to structural and functional anomalies in the frontal-subcortical circuits of the brain
(2) . Although the details of schizophrenia’s pathophysiology remain to be worked out, current evidence indicates that it is a complex disorder influenced by genes, environmental risk factors, and their interaction
(3) .
Over the past four decades, researchers have become increasingly certain that schizophrenia (Mendelian Inheritance in Man number 181500) has a complex multifactorial etiology
(4 –
7) . Among the relatives of patients, the risk for psychosis is about 50% of that of monozygotic twins—10% for first-degree relatives and 2% for second-degree relatives
(3) . A review of twin studies estimated the heritability of liability to schizophrenia to be 81%
(8) . And adoption studies show that schizophrenia is transmitted through biological rather than adoptive relationships
(9 –
11) . Despite this compelling evidence that genes influence susceptibility to schizophrenia, genome-wide linkage analyses of schizophrenia have produced conflicting results
(12) . Each of these linkage studies has identified regions showing some evidence for linkage to schizophrenia, but no finding has been consistently replicated.
One reason for conflicting results among prior studies is the use of relatively small samples. Risch and Merikangas
(13) showed that linkage studies have low power for complex diseases where multiple genes of small effect lead to disease, and Suarez et al.
(14) showed that, for genes of small effect, positive findings from small studies are unlikely to be replicated unless the replication sample is much larger. These considerations suggest that one approach to the difficulty of finding genes for schizophrenia is to collect a very large linkage sample of relatively homogeneous ethnicity using one set of methods. The present work took that approach and ascertained 557 Han Chinese families comprising 1,207 affected members. For comparison, the 20 schizophrenia linkage samples reviewed by Lewis et al.
(12) had a mean of 60 families and 147 affected members.
Method
Probands were recruited from six data collection field research centers throughout Taiwan. A detailed description of methods is given by Hwu et al.
(15) . To be included in the study, the family must have had two siblings with schizophrenia. We only included families of Han Chinese ancestry. Each proband underwent a diagnostic screen by a research psychiatrist using supplemental medical records and a semistructured interview that was based on DSM-IV
(16) .
Following this screen, the Mandarin Chinese version of the Diagnostic Interview for Genetic Studies
(17 –
19) was administered. We used the Diagnostic Interview for Genetic Studies, which was created by two of the investigators (M.T.T. and S.V.F.) and colleagues from the NIMH Human Genetics Initiative because it had been recommended for use in the NIMH Request for Applications that funded the work. The Diagnostic Interview for Genetic Studies makes a detailed assessment of the course of illness and a careful assessment of substance abuse and mood symptoms. This allows for an easier differential diagnosis between schizophrenia and 1) substance-related psychoses, 2) schizoaffective disorders, and 3) psychotic mood disorders. This detailed assessment helps protect against false positive diagnoses. Nurnberger et al.
(18) completed a reliability study of the Diagnostic Interview for Genetic Studies. Reliability for all individual diagnoses ranged from 0.73 to 0.95, except for schizoaffective disorder (0.29 and 0.49 for depressed and bipolar subtypes, respectively). Subsequent psychometric work by Faraone et al.
(17) showed 1) that the low kappa was due to its low base rate, 2) that it was frequently confused with schizophrenia (but not with bipolar disorder), and 3) that it had low sensitivity but high specificity (which would not create false positive ascertainments).
The Diagnostic Interview for Genetic Studies interviewers were college graduates who had had a major in psychology or psychiatric nursing and, on average, clinical experience with psychiatric patients for 2 to 3 years. All of the Diagnostic Interview for Genetic Studies interviewers received a 4-week training course, including lectures on psychopathology, DSM-IV diagnostic criteria, practice interviews with psychiatric outpatients, and practice interviews with psychiatric inpatients. Six sessions of videotaped reliability testing were completed before participation in the data collection. We checked their ratings of selected Diagnostic Interview for Genetic Studies items from major sections (e.g., depression, mania, psychosis). We discussed any discrepancy with the trainees. If a trainee made any mistake in any axis I diagnosis or more than three errors in individual items, he or she was required to continue training.
We supplemented structured interview data with medical records and a semistructured itemized assessment of psychopathology in family members called the Family Instrument for Genetic Studies. Interviewers underwent rigorous training on the Diagnostic Interview for Genetic Studies and the Family Instrument for Genetic Studies to ensure an accurate diagnostic assessment. Best-estimate final diagnoses were made by two board-certified research psychiatrists independently based on all the clinical information that was collected. When these psychiatrists disagreed, a third diagnostician (H.-G.H.) resolved the disagreement by reviewing all data schedules and medical records, and if necessary, a discussion with the field psychiatrist who cared for the patient would be arranged for clarification of the clinical condition. For all subjects, the minimal set of information required to make a diagnosis comprised the Diagnostic Interview for Genetic Studies, the Family Instrument for Genetic Studies, and information from medical records. In addition, for each proband, we required data from the structured psychiatric screening interview. When diagnosing schizoaffective disorder, DSM-IV requires that “symptoms that meet criteria for a mood episode are present for a substantial portion of the total duration of active and residual periods.” We operationalized “substantial portion” as one-third or more, with the mood symptoms contributing to the functional impairments of the patient.
Genotyping was conducted by the Center for Inherited Disease Research, with 386 markers spaced at an average of 9-cM intervals, following the center’s standard genotyping procedures (http://www.cidr.jhmi.edu/protocol.html). The discordancy for duplicate genotypes assayed on the same plates as the study genotypes was 0.06%. Marker distances were generated by using the sex-averaged Marshfield genetic map
(20) . Mendelian inconsistencies were checked with the Genetic Analysis System, version 2.0
(21) . RELCHECK was used to check for pedigree inconsistencies
(22) . The overall number of genotype and family errors was 0.39%.
Eleven discrepancies were found in six families. In two families, members thought to be full siblings were not and were excluded. In one family, a sibling was incorrectly coded as a parent, and the relationship was changed accordingly. Two individuals were found to be monozygotic twins, and therefore they were removed from the analysis. In one family, two parents were likely to be related by two or more degrees, so the family was excluded. The data reported here were collected through NIMH’s genetic initiative for schizophrenia. The genotypes and DNA samples are now publicly available and can be obtained through a formal request to NIMH. Clinical data are also available to the public. Details about how to access these data can be found at the following website: http://zork.wustl.edu/nimh.
Multipoint nonparametric genetic linkage analysis was performed on affected sibling pairs using the computer program MERLIN
(23) (because of the study design, only sibling pairs were available). Siblings were analyzed simultaneously to assess allele sharing by descent. We report the nonparametric linkage z scores calculated from MERLIN described by Whittemore and Halpern
(24) . Nonparametric linkage z scores were calculated at all markers and at 1-cM increments throughout the genome. Allele frequencies were calculated in MERLIN by using all available individuals. In the analysis reported here, only individuals with schizophrenia were considered affected. When this analysis was performed with consideration of both individuals with schizophrenia and schizoaffective disorder as affected, the results were virtually identical to those presented here. Empirical simulation studies evaluated the genome-wide significance of our findings using MERLIN. MERLIN generates simulated chromosomes that have the original data structure with regard to marker informativeness, marker spacing, and missing data patterns. Data are generated under the null hypothesis of no linkage and no association to our phenotype—schizophrenia. One thousand simulations were generated to determine genome-wide significance.
Discussion
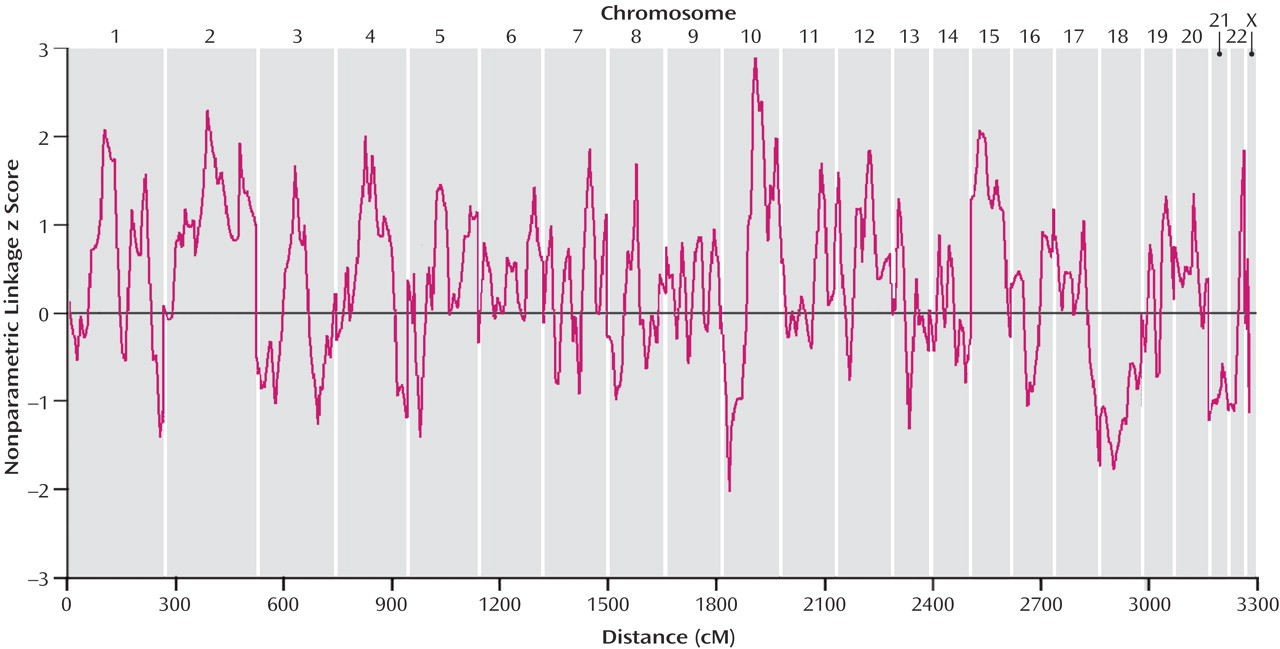
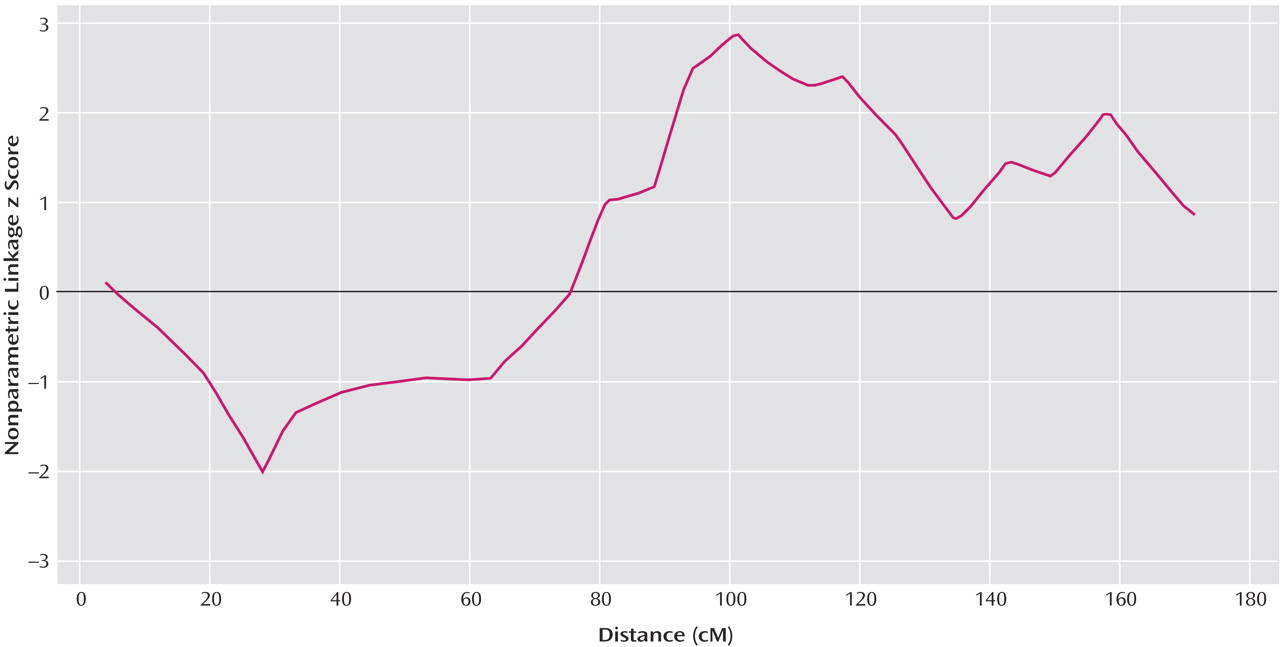
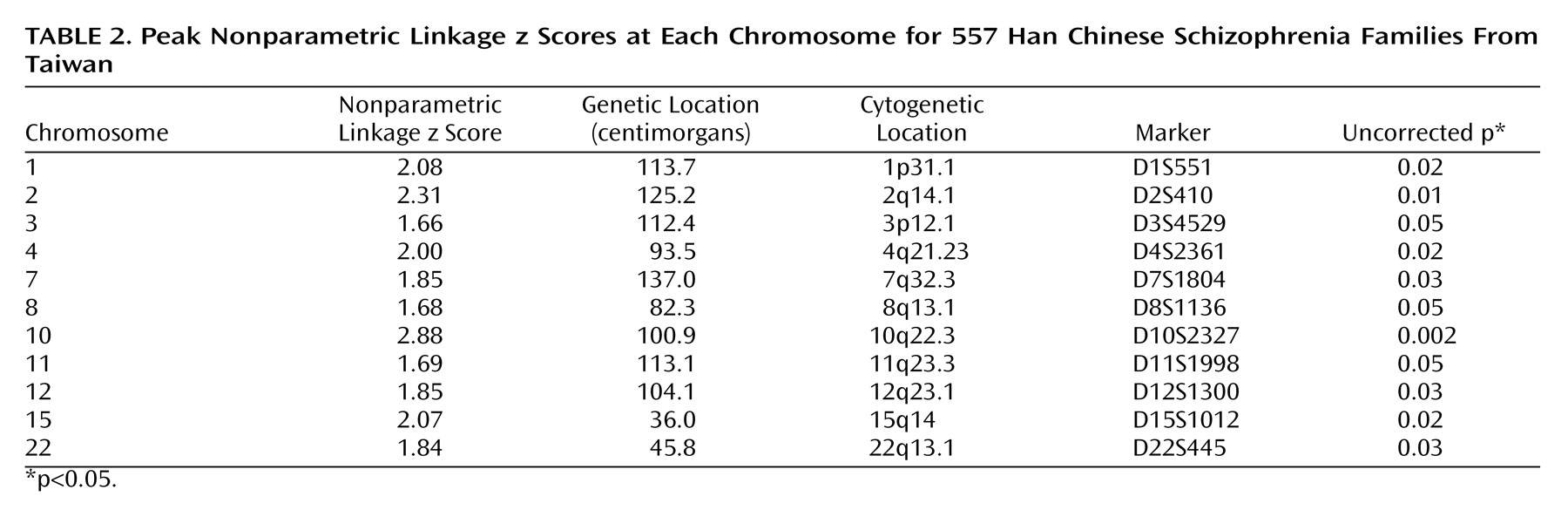
Our genome scan of Han Chinese families from Taiwan found evidence for a schizophrenia susceptibility gene at chromosome 10q22.3. Although our highest nonparametric linkage z score of 2.88 at 100.9 cM on chromosome 10 did not attain genome-wide significance, it provided statistically significant evidence for confirmed linkage, which was defined by Lander and Kruglyak
(25) as achieving p<0.01 for a locus meeting criteria for genome-wide significant linkage in a prior study. For our chromosome 10 finding, it was the prior study of genome scanning of Ashkenazi Jewish families by Fallin et al.
(26), which found a maximum nonparametric linkage score of 4.27 (p=0.00002) near marker D10S1744 at 107.2 cM on chromosome 10. The 95% CI for their region was 12.2 Mb, ranging from D10S1677 at 100.1 cM to D10S1753 at 112.6 cM. Considering that our finding was within this region, it seems reasonable to consider it as meeting the criteria of Lander and Kruglyak
(25) for confirmed linkage. Although the convergence of linkage data on 10q is noteworthy because we did not attain genome-wide significance, firm conclusions regarding linkage of this region to schizophrenia must await fine-mapping efforts that may increase or decrease the evidence for genetic linkage.
In contrast to our apparent confirmation of the finding of Fallin et al.
(26), two independent meta-analyses of schizophrenia linkage studies have not supported linkage to 10q22.3. Because the report by Fallin et al.
(26) was completed subsequent to the meta-analyses, it did not contribute to the evidence for linkage. The multiple probability scan meta-analysis of 18 genome scans of Badner and Gershon
(27) identified loci on chromosomes 8p, 13q, and 22q as showing strong evidence for linkage to schizophrenia. Other promising regions were 1q, 2q, 6q, and 15q, but evidence for linkage at these loci was weaker, indicating a need for further replication. Lewis et al.
(12) applied genome scan meta-analysis to 20 schizophrenia genome scans. They found statistically genome-wide significant evidence for linkage to chromosome 2q. Their results also suggested the following regions to be strong candidates: 5q, 3p, 11q, 6p, 1q, 22q, 8p, 20q, and 14p.
Our second largest finding was on chromosome 2q at 125.2 cM, which is located in the significant region reported by Lewis et al.
(12) . DeLisi et al.
(28), Levinson et al.
(29), and Moises et al.
(30) all reported this region among their largest results, and positive findings were also reported by Coon et al.
(31) and Straub et al.
(32) . Although these findings represent a broad region of chromosome 2, this area has consistently shown modest positive linkage findings. The linkage analysis in this article, which is one of the largest to date, also shows modest results in this region, suggesting that further, more detailed study is merited. Although our finding on chromosome 11 was modest, this region is ranked as the third strongest finding in the Lewis et al. meta-analysis. Also of note is our finding on chromosome 1, which is located in the region adjacent to the significant linkage reported by Brzustowicz et al.
(33) and supported by Gurling et al.
(34) .
We considered the possibility that the differences between our study and the conclusions of the meta-analyses were due to our use of a Han Chinese sample. The possibility of disease gene heterogeneity across ethnic groups has not been systematically addressed in genetic linkage studies of schizophrenia. Because gene frequencies vary among racial groups, some functional polymorphisms may be more predominant in one group than another. And because of interactions with other genes or environmental risk factors, the same gene variant can have different functional consequences across racial groups.
Ethnic differences could also reflect systematic differences between the environments of racial groups in events such as obstetric complications, which are known to be risk factors for schizophrenia
(35,
36) . If a specific risk factor, such as anoxia, interacts with a specific susceptibility allele, then we would expect to find greater evidence of linkage for the race that has the greatest prevalence of anoxia. Such effects have been hypothesized to explain the increased prevalence and sibling concordance for schizophrenia among African Caribbeans in the United Kingdom
(37) .
Despite these considerations, there are several reasons why ethnicity is unlikely to account for our results. When examined separately, none of the prior studies showed evidence for linkage to 10q22.3, so it is not the case that the meta-analyses obscured a few significant findings at that locus. Fallin et al.
(26) studied a sample of Ashkenazi Jewish families, and we studied Han Chinese families. Because we know of no clinical or epidemiological features of schizophrenia unique to these two ethnicities or any special population genetic relationship, it seems unlikely, a priori, that they would share a schizophrenia susceptibility gene not seen in other populations.
Other studies of Chinese samples argue against racial heterogeneity as an explanation for our results. None of these studies found evidence for linkage to 10q22.3
(38 –
42), but several found evidence of linkage to regions implicated by European-American samples. Examples include evidence by Hwu et al.
(41) of linkage to 1q22-q31, the finding by Li et al.
(40) at 22q11, the evidence of Hwu et al.
(39) supporting linkage to 6p22-p24, the linkage evidence of Liu et al.
(42) for 8p21, and the support of linkage for 15q13-14 by Liu et al.
(38) .
Our work should be viewed in the context of several limitations. The Diagnostic Interview for Genetic Studies interviewers had only 2 to 3 years of clinical experience. Although the Diagnostic Interview for Genetic Studies had been designed for use by interviewers with this level of training, the use of more experienced interviewers would have been better. This problem was also mitigated by the fact that all probands underwent a diagnostic screen by a research psychiatrist using supplemental medical records and a semistructured interview. Although our sample was very large, it may still not have sufficient power to detect susceptibility genes for schizophrenia in the presence of clinical and genetic heterogeneity, gene-environment interaction, and polygenic transmission.
The main strength of our study is its very large size. We collected 557 families comprising 1,207 affected members. In contrast, the 20 samples presented in the meta-analysis by Lewis et al. had a mean of 60 families and 147 affected members. Given this very large sample size, it is surprising that we did not find evidence for linkage to regions that have been implicated by prior studies. Although this may indicate that susceptibility regions implicated by prior studies are not relevant to the Han Chinese population, it is also possible that evidence for linkage was diminished by the main limitation of our study, i.e., the relatively low information content of the markers. Further work with a dense marker map is needed to evaluate this hypothesis.
Our results emphasize both the strengths and weaknesses of the linkage study method for schizophrenia. We have shown that a large sample can produce a result consistent with prior work but have also failed to replicate other findings. Thus, despite the progress achieved by our work and prior studies, all of this work was limited by the low power of linkage analysis for the finding of genes of small effect, a fact that has been previously noted
(13) .