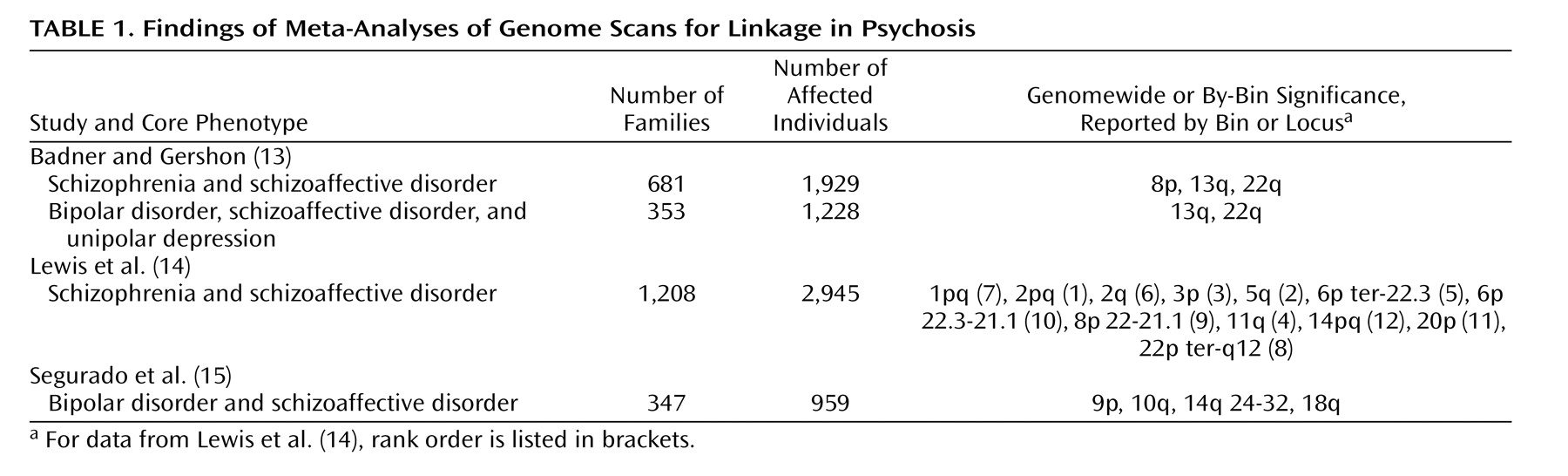
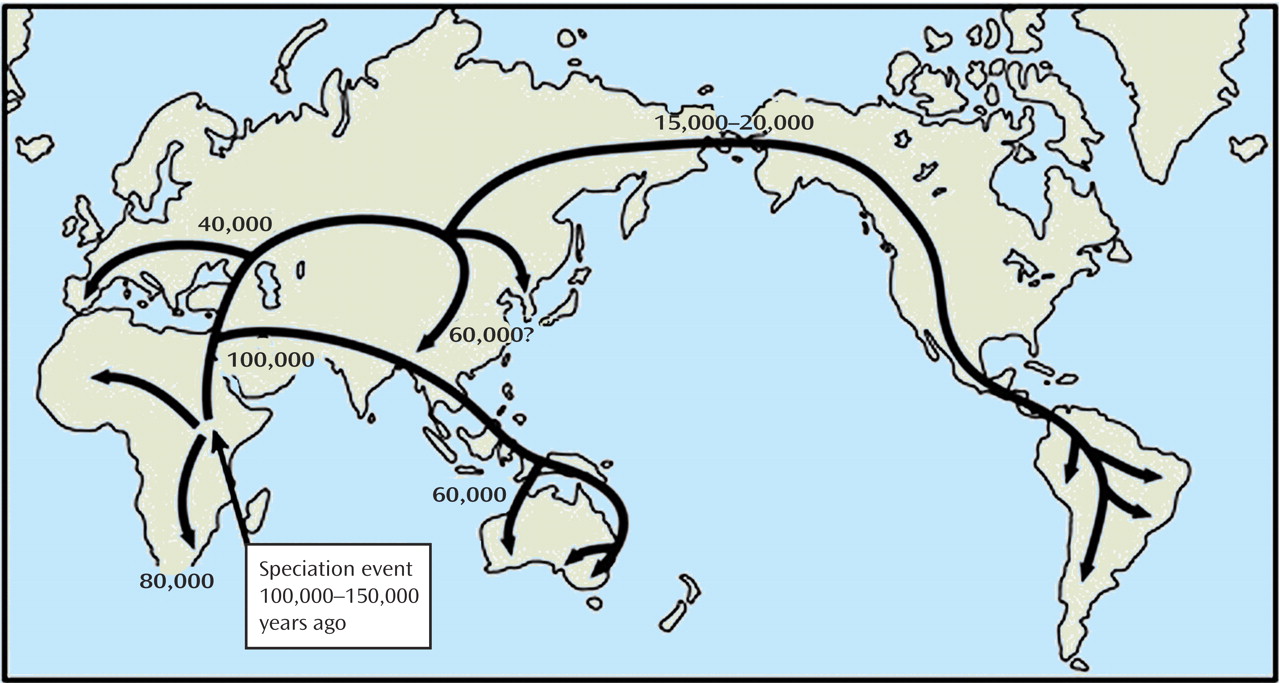
According to the Out of Africa hypothesis
(40), all modern human populations have an origin in Africa sometime between 100,000 and 150,000 years ago, at which time a species arose that replaced prior hominid species such as
H. Heidelbergensis,
H. erectus, and the Neanderthals (
Figure 2 ). This species had the capacity to represent the environment in symbols, a capacity manifest in language that can be traced back in the archaeological record through artifacts to 90,000 years ago, but not much earlier
(41,
42) . It is parsimonious to assume that the transition to language and the origin of the species reflect the same genetic event. To judge by the relative stability of the pattern of artifacts associated with preceding hominid species, the transition was relatively abrupt.
What can this genetic event have been? Broca
(43), following Dax
(44), reported that language production in the frontal lobes was located on the left side. In 1877 he wrote, “Man is, of all the animals, the one whose brain in the normal state is the most asymmetrical. He is also the one who possesses the most acquired faculties. Among these faculties . . . the faculty of language holds pride of place. It is this that distinguishes us most clearly from the animals”
(45) .
That Broca was right in supposing that directional asymmetry is the critical factor is now supported by studies of handedness in chimpanzees
(46) and other primates
(47) and studies of minicolumn separation in the planum temporale
(48) . Directional asymmetries are absent in other primates, including the great apes, although there is some evidence that tool use in
H. erectus was preferentially right-handed
(49) .
Thus, to understand the human brain, it is necessary to understand the genetic basis of asymmetry (and its anatomical correlate—the cerebral “torque” from right frontal to left occipital). Two theories of handedness
(50,
51) postulate a single gene acting together with a random factor. Although both originally predicted an autosomal location, there is an association between handedness and sex within families
(52) that is consistent with the presence of the gene on both X and Y chromosomes. Earlier, a gene in a region of X-Y homology was suggested on the basis of observations of sex chromosome aneuploidies
(53), and this is supported by intergenerational evidence from family studies
(54,
55) . The question remains: How could an apparently new feature—in this case cerebral asymmetry—arise from the existing genome complement? One hypothesis, championed, for example, by Goldschmidt
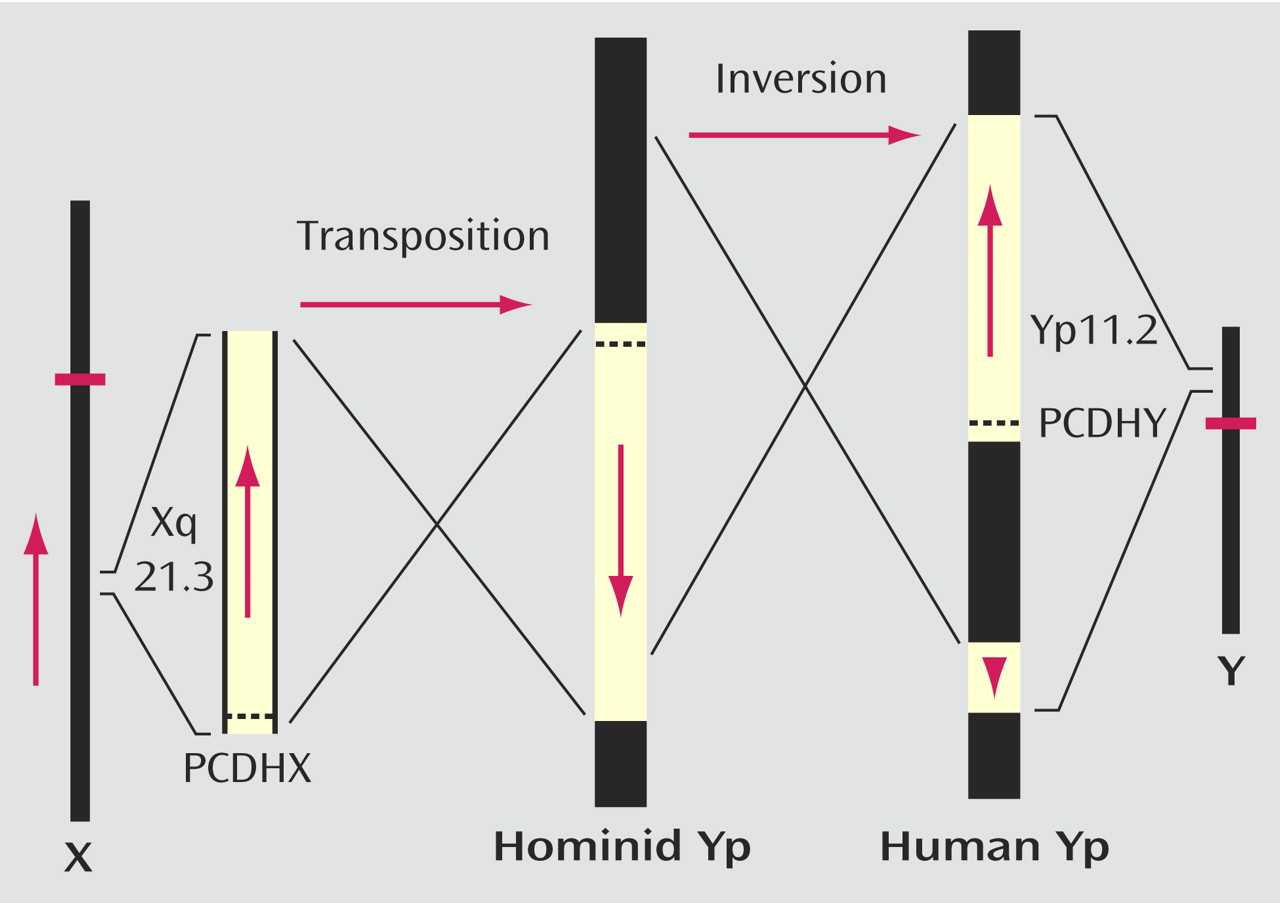
(56), an advocate of saltational change in evolution, is that chromosomal rearrangements play a role. Have there been chromosomal changes that might be relevant? One such change is the X to Y translocation followed by a paracentric inversion on the Y short arm that created a region of homology between Xq21.3 and two blocks on Yp after the separation of the chimpanzee from the hominid lineages
(57 –
59) . These rearrangements ensured that gene sequences that are present on the X in all mammals are present in man on the Y chromosome also. The direction of the sequence on the Y was reversed in the original translocation (estimated to have taken place approximately 6 million years ago—that is, close to the time of, and maybe the cause of, separation of the chimpanzee and hominid lineages) and in the paracentric inversion was reversed again in the distal block in Yp (
Figure 3 ). Because the arrangement of the homologous regions on the Y is universal in modern human males, these changes must have been selected (i.e., the gene content of the block duplicated on the Y is relevant), and the chromosomal changes constitute speciation events in hominid evolution. The first rearrangement is a possible correlate of the transition from the chimpanzee-hominid precursor to
Australopithecus, and the second (the paracentric inversion which cannot yet be dated) is a possible correlate of the transition to modern
H. sapiens . Within this region of homology, a gene pair (protocadherin X/Y) coding for cell surface adhesion molecules has been identified
(60) that has been subject to accelerated evolution (16 amino acid changes in the Y and five in the X sequence) in the hominid lineage by contrast with the relative stability of the sequence on the X in earlier primate evolution. This gene pair therefore is a candidate determinant of cerebral dominance for language.
Of particular interest is the status of protocadherin X/Y (and other genes within this region) with respect to X inactivation. Genes that are also present on the Y chromosome are protected from this epigenetic process, a protection that can be understood as aimed at achieving gene dosage equivalence in males and females. Therefore, whereas in all other mammals protocadherin X may be expected to be inactive on the inactive X chromosome, in females in
H. sapiens a new situation pertains. The mechanism by which protection from X inactivation is brought about is obscure, but it must be supposed to depend on an interaction between X and Y chromosomes, and this can only take place in male meiosis
(61) . If it depends on pairing of X-Y homologous sequences, then clearly the direction of the sequence on the Y and therefore the paracentric inversion will have significance. Whatever influence the presence of protocadherin Y may have had on the inactivation status of protocadherin X before the paracentric inversion, that influence will have been greater after this event. Thus, the orientation of the gene sequence on the Y must be supposed to have a key influence on the balance of gene expression in the two sexes in modern
H. sapiens relative to earlier hominid species.