Huntington’s disease is an autosomal dominant neurodegenerative disorder with principal symptoms of choreiform movements, psychiatric symptoms, and cognitive decline. Psychiatric disturbances are very common in Huntington’s disease, with frequencies of 35%–75% reported
(1–
3). An increased association of schizophrenia-like psychosis with Huntington’s disease has been documented
(4–
7). Prevalence estimates of schizophrenia-like psychosis in Huntington’s disease patients range from 5% to 16%
(8), much higher than the prevalence of 1% for schizophrenia in the general population. However, the reasons for this association are unclear.
The Huntington’s disease gene is located on chromosome 4p16.3 and contains an elongated and unstable CAG repeat
(9). In unaffected individuals, the normal gene contains 10–27 CAG repeats, which code a polyglutamine protein. In affected individuals, the number of repeats is expanded to greater than 36 CAGs. Previous studies have shown an inverse relationship between the number of CAG repeats and age at onset of Huntington’s disease, with a higher number of repeats associated with earlier age at onset
(10–
13). This relationship is the strongest in patients with juvenile-onset Huntington’s disease (<20 years old), who typically have the largest number of repeats (>60 CAG repeats)
(13). However, the relationship between the number of repeats and other phenotypic manifestations has not been as clearly demonstrated. In particular, there is no apparent association between the number of CAG repeats and the presence of behavioral and psychiatric symptoms
(14–
16).
Several investigators have reported that a subset of families in which Huntington’s disease occurs have a predisposition for developing psychosis
(17,
18). We have previously reported the clinical characteristics and genotypes of two families in which Huntington’s disease occurs, one with a juvenile Huntington’s disease proband with psychotic features and the other with a juvenile proband without psychosis
(19). Comparisons of multiple demographic and clinical characteristics did not show significant differences between the two probands. In fact, the number of CAGs in the psychotic Huntington’s disease proband was lower than in the nonpsychotic proband (68 versus 81 repeats). However, the father and paternal grandmother of the proband with psychosis also had Huntington’s disease and had been psychotic early in the course of their illness. The psychotic proband met the criteria for a DSM-IV diagnosis of schizophrenia (with the exception of the criterion “substance/general medical condition exclusion”) on the Structured Clinical Interview for DSM-IV Axis I Disorders—Patient Edition (SCID-P)
(20). This preliminary study suggested that psychotic features in Huntington’s disease might be associated with the presence of family history of Huntington’s disease with psychosis.
In the current study, we extended our investigation to a larger group of Huntington’s disease patients with psychosis to address the following questions: 1) Is age at onset of psychosis related to the number of CAG repeats? 2) Is there an excess of psychosis in family members of Huntington’s disease patients with psychosis compared to that observed in the general population? 3) If so, is psychosis more likely to occur in family members with or without Huntington’s disease? The presence of psychosis only in those with Huntington’s disease (segregation of psychosis with the Huntington’s disease gene) would suggest that the psychotic features are a consequence of having Huntington’s disease and that psychosis is perhaps unmasked by the Huntington’s disease gene. On the other hand, if psychosis occurs in family members without Huntington’s disease as well as those with Huntington’s disease, there may be an unrelated gene or other pathological processes associated with the susceptibility of developing psychosis independent of Huntington’s disease.
Method
Huntington’s disease patients at the medical genetics clinics of the University of Washington in Seattle and the University of British Columbia in Vancouver, Canada, were eligible to participate in the study. Because this study was not a population-based study to evaluate the prevalence of psychosis, we identified patients with known behavioral problems through clinical databases. Overall, 80 Huntington’s disease patients with a history of behavioral problems were screened for inclusion (25 subjects from the University of Washington and 55 subjects from the University of British Columbia). To be included in the study, patients needed to have 1) a clinical diagnosis of Huntington’s disease; 2) sufficient information in their medical records to establish a reliable DSM-IV psychiatric diagnosis, including detailed descriptions of delusions, hallucinations, or bizarre behavior; and 3) available data on Huntington’s disease genotype. Medical and psychiatric records were reviewed by two board-certified psychiatrists (D.T., F.S.), and psychiatric diagnoses were established according to DSM-IV by the two psychiatrists.
Twenty-two subjects met the research inclusion criteria for Huntington’s disease and psychosis; the remaining subjects were excluded because their medical records had insufficient documentation of psychotic symptoms, even though some of them had been diagnosed as “schizophrenic.” The medical records of 33 Huntington’s disease patients without known psychosis who were evaluated during the same time period were reviewed to determine whether the patients were eligible for inclusion as nonpsychotic comparison subjects. Twenty-two patients who had detailed clinical records with no evidence of psychosis were included in the comparison group. The other 11 subjects were excluded from further analysis because their medical records were not sufficiently detailed. The nonpsychotic comparison subjects were not matched to the subjects with psychosis on any demographic or clinical characteristics.
The following information was extracted from subjects’ medical records: 1) demographic characteristics, including age, education, marital status, ethnic background, and employment history; and 2) clinical characteristics, including presence or history of and estimated age at onset of motor symptoms or chorea, cognitive decline, major or minor depression, substance use, suicidal attempts, persistent personality change, auditory or visual hallucinations, delusions, and functional impairment. Age at onset was defined as the age at which persistent motor, behavioral, or cognitive symptoms in the subject were first observed by family or health care providers.
Psychiatric diagnoses and identification of subjects as psychotic or nonpsychotic were confirmed in a subset of 24 subjects. Eight subjects with psychosis and 16 without psychosis provided informed consent (in accordance with the procedure established by the University of Washington human subjects review committee) and were interviewed with the SCID-P by D.T. and F.S. The two raters arrived at consensus diagnoses. Subjects who had been identified as psychotic on the basis of medical records met the criteria for schizophrenia, major depressive disorder, bipolar disorder with psychotic features, or dementia. Because all subjects had Huntington’s disease, the exclusion on the basis of a “general medical condition” in the DSM-IV criteria for the psychotic disorders was not applied. Subjects who had been identified as nonpsychotic on the basis of medical records did not meet DSM-IV criteria for any of the psychotic disorders.
Available medical records of subjects’ first-degree family members were reviewed, and data on the demographic and clinical characteristics described earlier in this section were extracted. The presence of Huntington’s disease (identified through documentation of Huntington’s disease genotype or autopsy findings) or psychosis in the family members was determined through review of their medical records. If psychosis was not reported, we presumed that psychosis was not present. This criterion was used consistently for relatives of both psychotic and nonpsychotic subjects.
Statistical Analysis
Chi-square tests or chi-square tests with Yates’s correction were used for comparison of categorical variables. Unpaired two-sample t tests were used for comparison of continuous variables. The Pearson product-moment correlation was used for bivariate correlation of continuous variables. For the analysis of family history of psychosis, we did not have access to enough medical records for subjects’ second-degree relatives to consistently assess the presence of psychosis in those relatives. Therefore, only subjects who had a first-degree relative with documented psychosis were considered to have a positive family history of psychosis. A total of 20 families in which the proband was psychotic and 21 families in which the proband was nonpsychotic were included in the analysis of family history. Families that included more than one subject were counted once in the analysis of family history, and one family was omitted from the analysis because the subject was adopted and the family history was unknown.
Huntington’s Disease Genotyping
For each subject, the Huntington’s disease gene containing the CAG repeat was amplified, and the number of repeats was determined and compared with that of sequenced clones of known size
(21,
22) at the University of British Columbia medical genetics laboratory. The normal (asymptomatic) number of CAG repeats is 6–35, and the number in affected individuals is >35. The measurement of CAG repeats allows direct identification of the mutation that is causative in Huntington’s disease
(23).
Results
The study subjects were 44 patients with Huntington’s disease, 22 of whom had psychotic symptoms and 22 of whom had no psychotic symptoms. The diagnosis of Huntington’s disease was confirmed in all patients by means of Huntington’s disease genotyping and clinical examination. Of the 22 psychotic probands, eight had psychosis only, eight had mixed affective and psychotic features, and six had psychotic symptoms in the context of dementia. Among the 22 nonpsychotic comparison subjects, major depressive disorder was the most common psychiatric diagnosis (N=10), followed by alcohol use disorder (N=6), other substance use disorders (N=4), adult antisocial behavior (N=1), bulimia nervosa (N=1), personality change due to Huntington’s disease (N=3), and dysthymic disorder (N=2). (The numbers total >22 because some subjects had more than one diagnosis.) Four comparison subjects did not meet the criteria for any DSM-IV diagnosis.
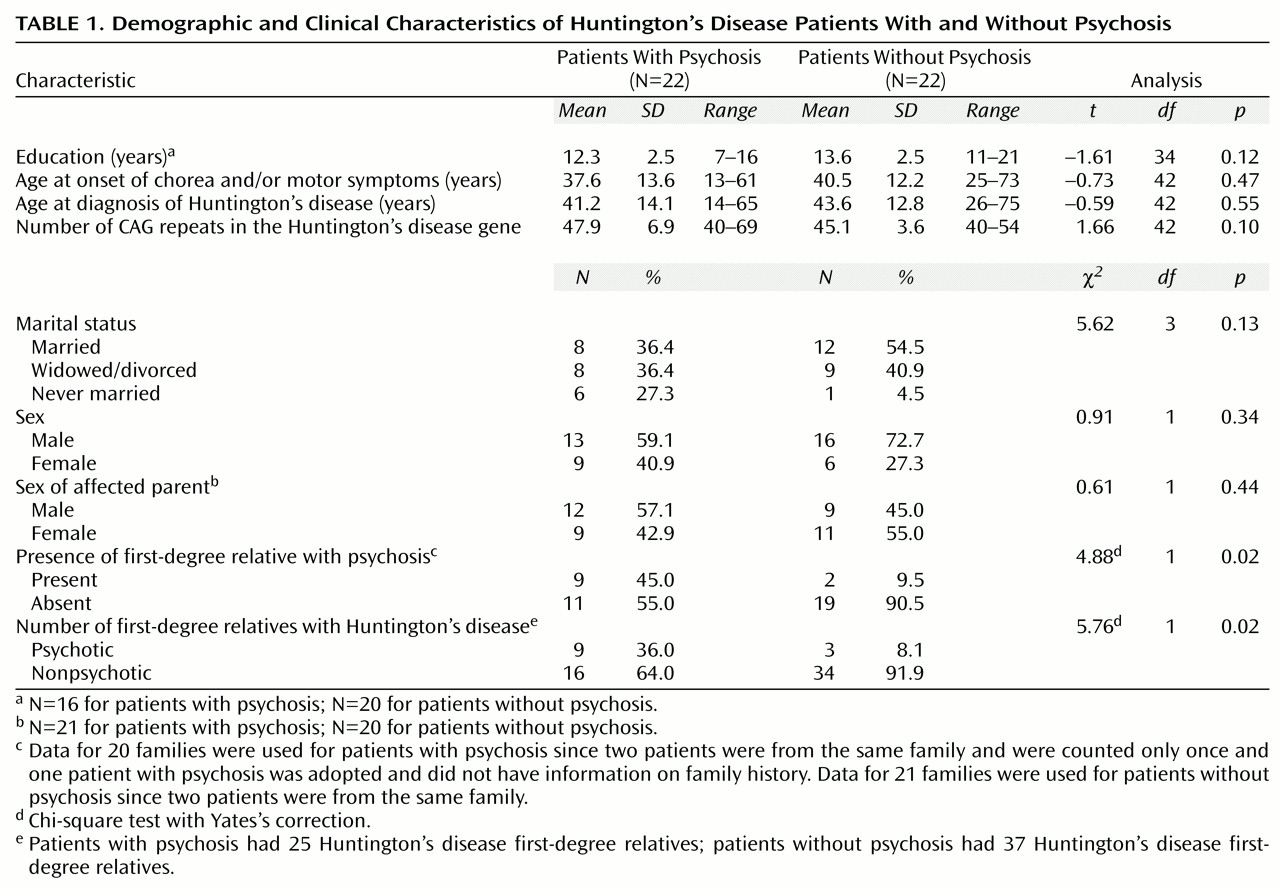
Comparisons between probands with and without psychotic symptoms showed no significant differences in demographic and clinical characteristics (
Table 1). The probands with psychotic symptoms had less education and a higher number of CAG repeats than the comparison group, although the differences were not statistically significant (two-sample t tests, t=–1.61, df=34, p=0.12, and t=1.66, df=42, p=0.10, respectively). Probands who had psychotic symptoms were much more likely to have a first-degree relative with psychosis than were the nonpsychotic comparison probands (Yates’s χ
2=4.88, df=1, p=0.02). In addition, psychosis was highly associated with Huntington’s disease. Of the nine probands with psychosis who had first-degree relatives with psychosis, only one proband had a first-degree relative with psychosis who did not also have Huntington’s disease. The 20 families of probands with psychosis included nine first-degree relatives with both Huntington’s disease and psychosis as well as 16 first-degree relatives with Huntington’s disease who did not have psychosis.
The 21 families with nonpsychotic probands included three first-degree relatives with both Huntington’s disease and psychosis and 34 first-degree relatives with Huntington’s disease who did not have psychosis. The three psychotic Huntington’s disease relatives were related to two nonpsychotic probands; one proband met the DSM-IV criteria for personality change secondary to Huntington’s disease, and the other met the criteria for alcohol use disorder and other substance use disorder. Comparison of the number of relatives with both Huntington’s disease and psychosis versus the number with Huntington’s disease but no psychosis by proband status (psychotic versus nonpsychotic) showed a significant difference (Yates’s χ2=5.76, df=1, p=0.02). The estimated risk of developing psychosis for first-degree relatives with Huntington’s disease was 36% for relatives of probands with psychosis and 8% for relatives of probands without psychosis.
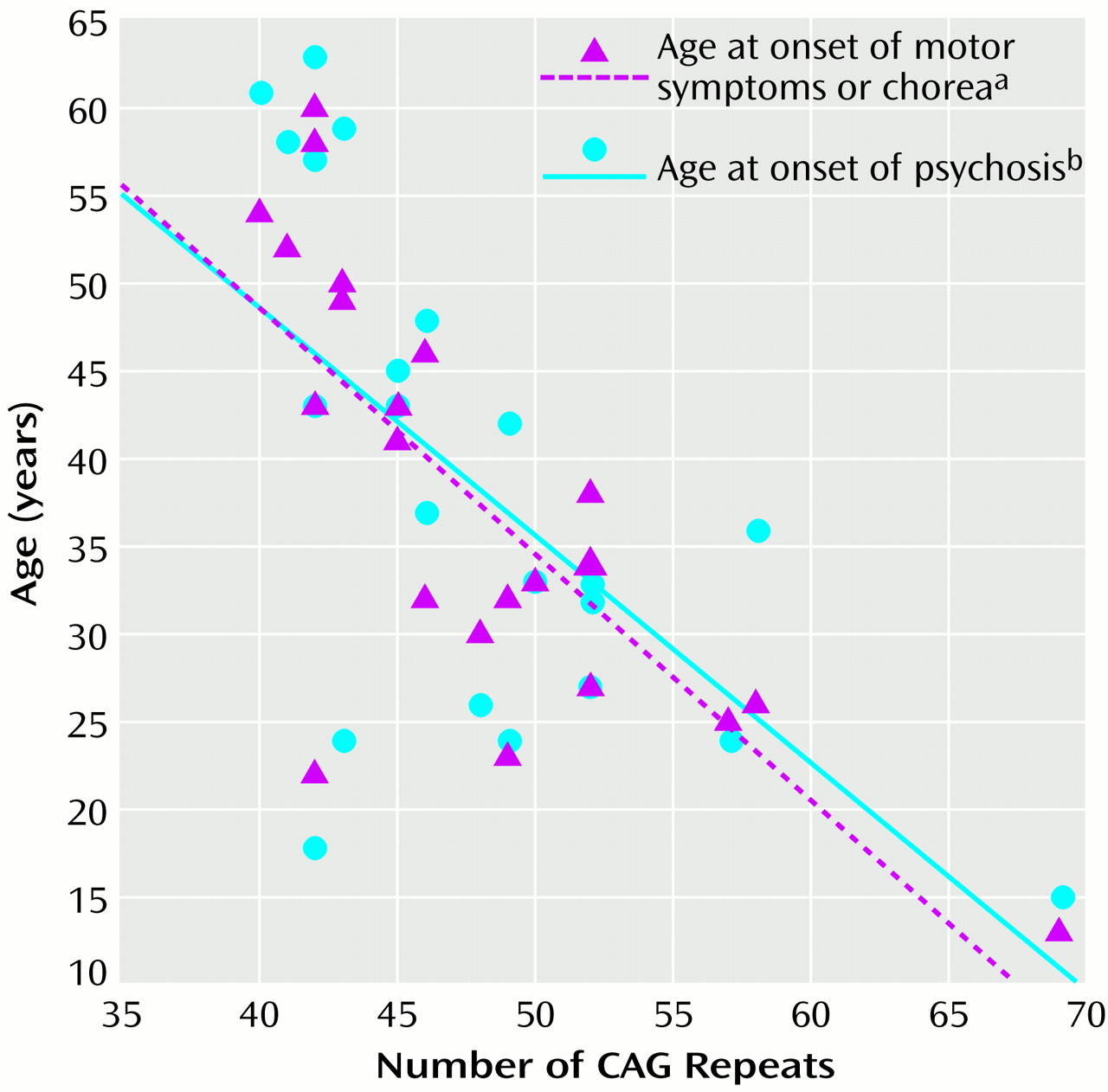
For the subjects with psychosis, the age at onset of psychosis correlated with the age at onset of Huntington’s disease motor symptoms or chorea (r=0.86, df=1, p
<0.0001). There was an inverse correlation between age at onset of psychotic symptoms and the number of CAG repeats, although the correlation between number of CAG repeats and age at onset of motor symptoms and chorea was higher (
Figure 1).
Discussion
Others have previously documented the higher than expected occurrence of psychotic symptoms in individuals with Huntington’s disease
(24,
25). Folstein et al.
(7) used systematic psychiatric interviews in a population-based study of 186 Huntington’s disease patients in Maryland and found that eight (4.3%) had received a diagnosis of schizophrenia over a 10-year period. Other population-based studies have estimated the prevalence of schizophrenia in Huntington’s disease to range from 0% to 12%
(4–
6,
26). The use of differing diagnostic criteria and subject selection procedures is likely to account for the variability in estimates. However, because the study reported here was not population-based, we cannot derive prevalence estimates of schizophrenia-like syndromes in Huntington’s disease. Nevertheless, there is a general consensus that the rate of occurrence of schizophrenia-like psychosis in Huntington’s disease is higher than what would be expected by chance alone, although the reasons for this association are unclear. Moreover, previous reports have suggested that some families in which Huntington’s disease occurs also have a higher occurrence of psychosis and other behavioral problems
(18,
19). Our findings support the hypothesis that psychosis is more likely to occur in some Huntington’s disease families because Huntington’s disease predisposes members of those families to develop psychosis rather than the hypothesis that some Huntington’s disease families carry additional “psychosis” genes.
Previous studies have found no significant differences in number of CAG repeats between Huntington’s disease patients with psychiatric or nonpsychiatric phenotypes
(13,
15,
16). However, the subjects with psychiatric phenotypes in these studies included subjects with depression, personality changes, and dementia as well as those with psychosis. We found that subjects with Huntington’s disease and psychosis had a higher number of CAG repeats than comparison subjects without psychosis, although the difference was not significant. The Huntington’s disease gene may be associated only with certain behavioral phenotypes, such as psychosis. The pathological mechanism(s) underlying Huntington’s disease is yet unknown; therefore, we cannot exclude the possibility that some other undiscovered change in the Huntington’s disease gene may be involved in the development of the psychotic phenotype. Alternatively, additional genetic factors may predispose certain Huntington’s disease families to develop psychosis. There may be modifying genes that interact with the Huntington’s disease gene to increase this susceptibility. Additional genetic studies are necessary to address these questions.
To our knowledge, our study of Huntington’s disease patients with psychosis and their families included the largest reported series of patients with psychosis for which Huntington’s disease genotypes were available. Multicenter collaboration was necessary to assemble a large enough study group, which was possible with access to two large university-based medical genetics clinics that had evaluated a large number of Huntington’s disease patients. In addition, excellent medical records for other family members were available, allowing us to establish psychiatric diagnoses in the relatives.
A weakness of this study is that we were unable to perform face-to-face interviews of all subjects and first-degree relatives. However, psychiatric diagnoses based only on medical records is often a reasonable proxy for best-estimate diagnoses derived by using direct interviews with the subject and family history interviews with reliable informants, as well as available medical records
(27). Since we used medical records review to evaluate the relatives of both psychotic and nonpsychotic subjects, the frequency of false-negative identification of psychosis in the relatives was probably approximately equal in both subject groups. Family studies that include direct interviews of relatives will be necessary to validate our findings. In addition, two of the Huntington’s disease patients who were initially nonpsychotic developed psychotic symptoms during the course of follow-up (within approximately 5 years), suggesting that the onset of psychosis in Huntington’s disease patients is variable. This finding is consistent with results previously reported by Watt and Seller
(28). They found that three of seven Huntington’s disease patients with schizophrenia-like symptoms had psychotic symptoms years before the onset of the neurological signs and symptoms of Huntington’s disease, two patients presented with concomitant psychotic and neurological symptoms, and the remaining two patients developed psychotic symptoms 4–12 years after the onset of motor symptoms or chorea. Future studies should take this variability into account and should include longer follow-up periods.
In conclusion, our findings suggest that the pathophysiological process underlying Huntington’s disease may contribute to the development of psychotic symptoms in a subset of patients. However, since not all Huntington’s disease patients or families are susceptible to developing psychosis, other predisposing factors may also exist. The complex pathogenetic mechanisms underlying psychiatric disturbances in Huntington’s disease remain unknown. Further neurobiological and genetic studies involving Huntington’s disease patients with psychosis are necessary to elucidate the underlying mechanism(s) of action.