Genetic factors are known to play the major role in the etiology of bipolar affective disorder, but the specific genes involved and their modes of inheritance remain unknown (for review, see reference
1). Family study data suggest that the inheritance of bipolar affective disorder is likely to be complex and multifactorial, with at least a few distinct genes acting together to cause the phenotype and with considerable etiologic heterogeneity
(2,
3).
Parent-of-origin effects may be an important part of this complexity. Previously, we reported excess maternal transmission of bipolar affective disorder in a set of multiplex pedigrees ascertained through a proband with bipolar affective disorder
(4). We observed more affected mothers than fathers, an increased risk of bipolar affective disorder for maternal versus paternal relatives of the probands, and several extended families with exclusively maternal transmission of bipolar affective disorder. Excess maternal transmission of bipolar affective disorder has since been observed in other multiplex family samples
(5,
6) but not in some unselected samples
(7,
8). Excess maternal transmission might reflect subtle ascertainment biases, imprinting, X-linked genetic factors, vertical transmission of infectious agents, in utero or other early maternal influences on offspring, or mutations of the mitochondrial DNA (mtDNA), which is transmitted only by mothers. Among these many possible mechanisms, mutations of the mtDNA are the most readily investigated by molecular methods.
The 16.5 kilobase circular mtDNA resides outside of the nucleus. The mitochondrial genome encodes proteins essential for oxidative phosphorylation (for review, see reference
9). Mutations in mtDNA are known to cause several maternally transmitted diseases. Many of these diseases show prominent neuropsychiatric symptoms, reflecting the essential role of oxidative phosphorylation in neuronal function. The mtDNA has also been well characterized at the population level, and several clusters of mtDNA variants (haplogroups) associated with particular populations have been described. The study reported here was designed to test the hypothesis that point mutations in the mtDNA play an etiologic role in bipolar affective disorder. We sequenced the entire mitochondrial genome in nine unrelated probands with bipolar affective disorder selected from families with strictly maternal transmission. Selected variants were tested for association with bipolar affective disorder in a sample of 92 unrelated probands and matched comparison subjects who were classified into the major European haplogroups. Our results show that bipolar affective disorder occurs across all of the major European mtDNA haplogroups but do not reveal any point mutations that explain excess maternal transmission of bipolar affective disorder.
Method
Sample Collection and Evaluation
Subjects were ascertained and evaluated as described previously
(10). Briefly, probands with at least two sibs or one sib and only one parent with a major affective disorder by family history were enrolled for the study. Subjects were interviewed by a psychiatrist using the Schedule for Affective Disorders and Schizophrenia—Lifetime Version
(11). Apparently bilineal families and those in which at least one putatively unaffected parent could not be interviewed were excluded. The interview data, along with family informant data and available medical records, were reviewed by two additional psychiatrists, who assigned a best-estimate diagnosis based on Research Diagnostic Criteria
(12). The assigned best-estimate diagnoses have high reliability (see reference
13). After complete description of the study to the subjects, written informed consent was obtained.
The nine unrelated probands selected for mtDNA sequencing included five men and four women. All self-reported exclusively European origins. Seven probands were assigned a best-estimate diagnosis of bipolar I disorder; two probands were given the diagnosis of bipolar II disorder with recurrent major depression. Age at first mania or major depression ranged from 15 to 35 years, with a mean of 24 years, typical of probands in this sample
(14). None of the probands reported a history suggestive of neurological disease or myopathy. Each proband belonged to a large multiplex pedigree in which at least one apparent maternal transmission but no apparent paternal transmissions of bipolar affective disorder were observed. Six of the nine pedigrees were described previously
(4).
The 93 probands selected for the case-control assay included 39 men and 54 women. Most (97.8%) reported exclusively European ancestry. All probands had been assigned a best estimate diagnosis of bipolar I (N=77; 82.8%), bipolar II (N=15; 16.1%), or schizoaffective-bipolar-manic (N=1; 1.1%) disorder, with an age at first mania or major depression ranging from 8 to 58 years (mean=21.4 years). Fewer data on extended pedigrees were available for these probands than for those selected for mtDNA sequencing, but by using the available data, the probands were grouped on the basis of the presence of affected relatives in the paternal or maternal lineages, as described previously
(4,
13). An apparent pattern of maternal transmission was seen in the families of 35 probands; an apparent pattern of paternal transmission was seen in the families of 23 probands. The remaining probands (N=35) had affected relatives on both sides or on neither side of their families.
The control group was composed of 63 paternal relatives of the probands (no more than one per family) who were past the age of risk for onset of bipolar affective disorder at the time they were interviewed. All had been determined to be free of mood disorders or related illnesses after best-estimate review of direct interview, family informant, and any medical record data. Paternal relatives were selected because they could not share mtDNA with the probands but would be better matched to the probands on environmental variables and nuclear genes than comparison subjects drawn from the overall population.
Sequencing
The probands’ mtDNA was sequenced in sets of three, using automated methods. Sets A (probands 55-001, 36-001, 25-005) and B (43-001, 184-001, and 291-001) were sequenced at Emory University. Set C (probands 381-001, 386-001, 322-001) was sequenced at Johns Hopkins University. All sequencing was performed by using a template that was amplified by means of polymerase chain reaction with Taq polymerase. Primer sequences and amplification conditions differed slightly between laboratories. (Details are available on request from the corresponding author.) Methods unique to each set of probands are briefly described below.
Set A
The entire mtDNA was amplified into 12 completely overlapping polymerase chain reaction fragments. These 12 fragments were purified using Centricon-100 microconcentrators (Amicon, Inc., Beverly, Mass.). Purified double-stranded polymerase chain reaction products were directly sequenced using the Taq DyeDeoxy Termination Cycle Sequencing Kit (Perkin-Elmer Applied Biosystems, Inc., Norwalk, Conn.). All sequencing reactions were then purified using Centri-Sep columns (Princeton Separations, Inc., Adelphia, N.J.) and run through 6% acrylamide/7 moles/liter urea gels on a 373A DNA Sequencer (Perkin-Elmer Applied Biosystems, Inc.). The resulting data were aligned using Sequencher 3.0 software (Gene Codes Corp., Ann Arbor, Mich.).
Set B
The entire mtDNA was amplified by polymerase chain reaction into nine partially overlapping fragments by using the primer pairs and amplification conditions described elsewhere
(15). Fragments were purified by using Centricon-100 microconcentrators. Purified double-stranded polymerase chain reaction products were directly sequenced in both directions by using the Big Dye Terminator Cycle Sequencing Kit (Perkin-Elmer Applied Biosystems, Inc.). All sequencing reactions were then purified using Centri-Sep columns and run through 5% Long Ranger/6M urea gels on a 377 DNA Sequencer (Perkin-Elmer Applied Biosystems, Inc.). The resulting data were aligned as for Set A above.
Set C
The entire mtDNA was amplified by using polymerase chain reaction into nine partially overlapping fragments using the primer pairs and amplification conditions described elsewhere
(15). Fragments were electrophoresed on a 1% Nusieve agarose gel and visualized by ethidium bromide. Appropriate sized bands were excised and purified by using Qiaquick Gel Extraction Kit (Qiagen, Valencia, Calif.). Purified polymerase chain reaction products were concentrated under vacuum by using Speed Vac (Global Medical Instrumentation, Inc., St. Paul, Minn.) to a concentration of approximately 6 ng/μl as determined by DNA Dipstick (Invitrogen Corp., Carlsbad, Calif.). Samples were sequenced in one direction at the Johns Hopkins Genetics Core Facility by using a 377 DNA Sequencer (Perkin-Elmer Applied Biosystems, Inc.), as described for Set B above.
Variant Typing
All variants were assayed by polymerase chain reaction amplification of the flanking region followed by digestion with a restriction endonuclease specific for either the variant or wild-type allele. Variants that did not involve an existing restriction site were assayed by changing the penultimate nucleotide of the reverse primer to create a restriction site in the presence of the variant. Whenever possible, polymerase chain reaction fragments were designed to contain a second, nonvariable restriction site as a positive control. Undigested and digested polymerase chain reaction products from sequenced mtDNA of a proband harboring the variant served as additional controls. Digests were subjected to agarose gel electrophoresis, stained with ethidium bromide or SYBR Green (Molecular Dynamics, Sunnyvale, Calif.), and visualized by using an ultraviolet transilluminator. The digest from one variant (nucleotide position 1888) could not be resolved on agarose, but it was easily resolved on a denaturing MDE gel (mutation detection enhancement gel solution) and visualized by silver staining (Amersham Pharmacia Biotech, Piscataway, N.J.). No variant showed evidence of heteroplasmy, but only substantial heteroplasmy would have been detected by these methods. Details of variant assays may be obtained on request to the corresponding author.
Haplotyping
Probands and comparison subjects were haplotyped for the major European haplogroups by using the primer pairs, amplification conditions, and digests described elsewhere
(16). Fourteen probands and nine comparison subjects could not be unambiguously assigned to a single haplogroup by these methods.
Statistical Analysis
The probability, P, of detecting a variant in a sample of sequences was calculated by using the equation, P=1–(1–
q)
n, where
q=frequency of the variant in the sample and
n=number of sequences compared (not including the “Cambridge” sequence, which is the published reference sequence
[17]). The power of our case-control panel to detect an association with a given variant was estimated from the standard equations for a case-control study
(18); alpha was set at 0.05.
Mean pairwise differences were calculated based on the total number of variant sites observed between pairs of sequences (including the Cambridge reference sequence) per 100 base pairs sequenced.
Frequencies were compared by using the chi-square test (with Yates’s correction). Scores were compared by using ANOVA; Tukey’s honest significant difference test was employed for specific comparisons. All p values were two-tailed. Multiple comparisons were subjected to Bonferroni correction.
Results
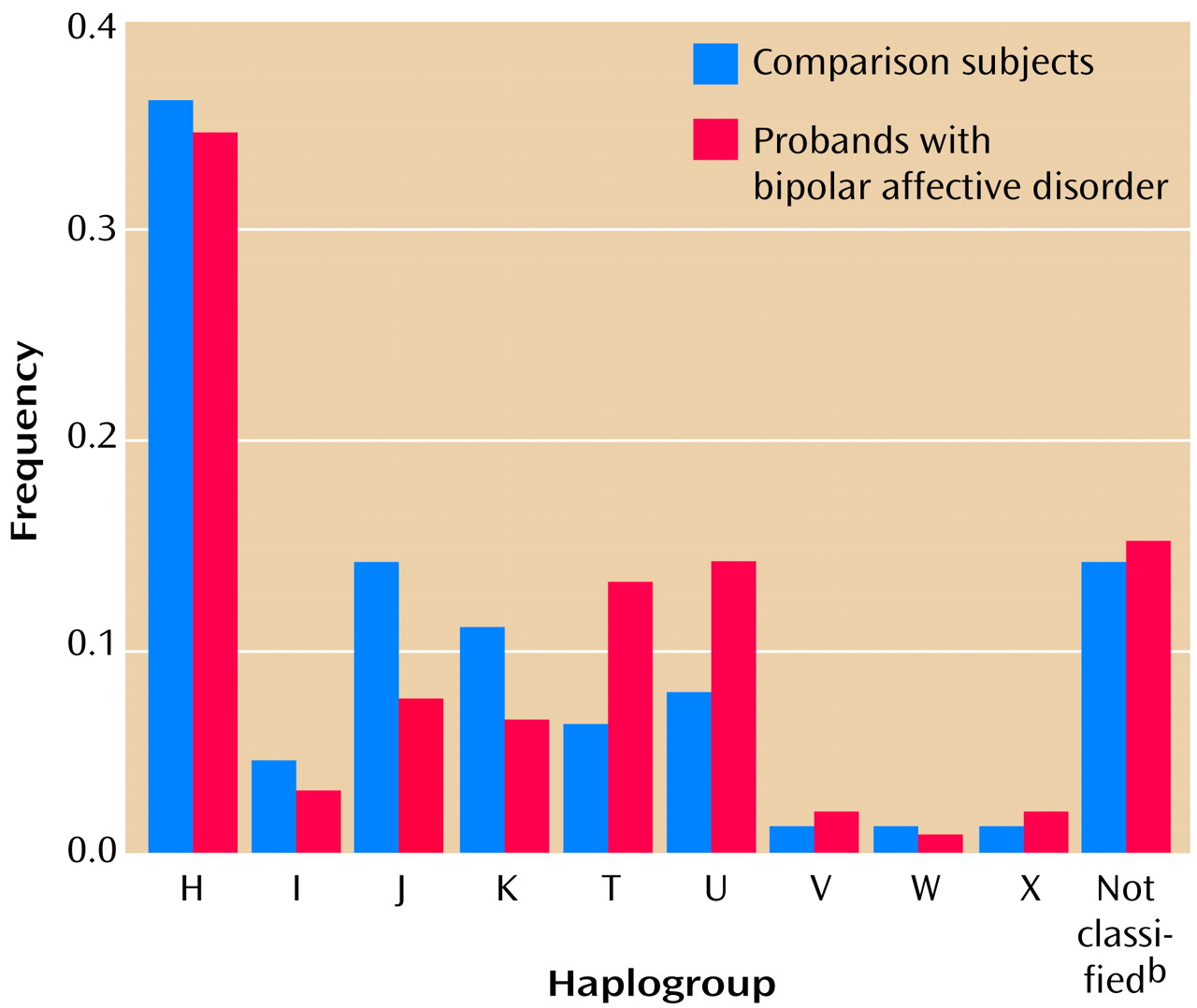
Haplotyping
The results of haplotyping for probands and comparison subjects are shown in
Figure 1. All of the major European haplogroups occurred among both probands and comparison subjects at the expected proportions (16, 19). No significant differences were found in haplogoup frequencies between probands and comparison subjects (overall χ
2=4.66, df=14, n.s.).
Sequencing
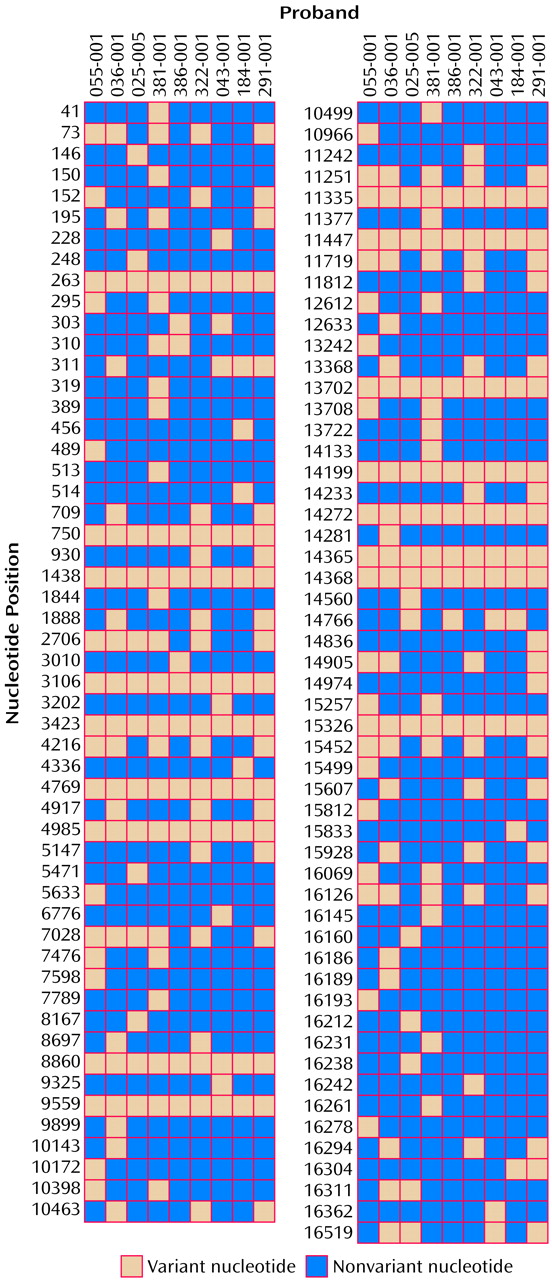
The results of the mtDNA sequencing for each of the nine probands with bipolar affective disorder are shown in
Figure 2. Compared to the Cambridge reference sequence
(17), 107 different sequence variants were observed. Sixteen variants, each found in all nine probands, probably represent errors or rare polymorphisms in the Cambridge reference sequence (20–24). Of the remaining variants, 21 would cause an amino acid change, 36 were synonymous (as they would not cause an amino acid change), 13 occurred in an rRNA or tRNA gene, and 37 occurred in untranslated regions.
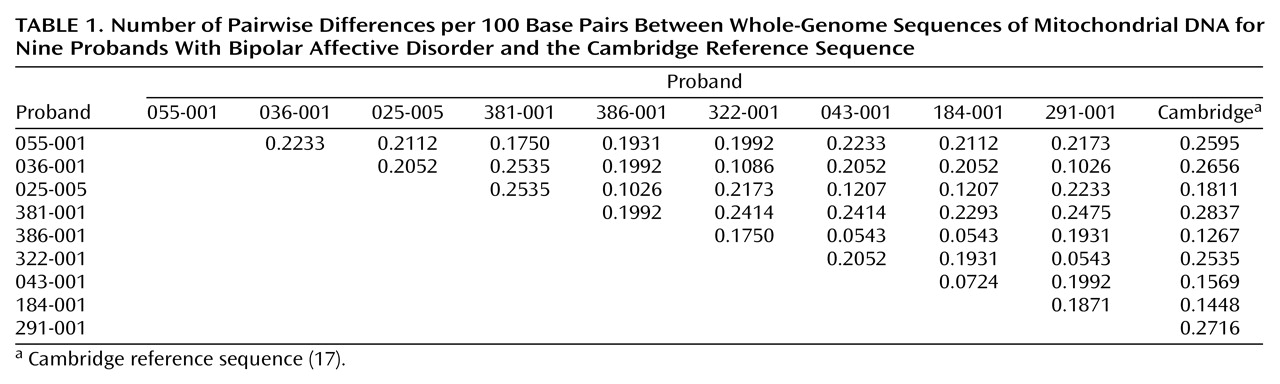
Considerable sequence diversity was observed in these nine probands. Each sequence was compared with every other sequence (including the Cambridge reference sequence) to estimate pairwise differences. The number of pairwise differences ranged widely, from 0.0543 to 0.2837 per 100 base pairs (
Table 1). The mean pairwise difference was estimated at 0.1880 per 100 base pairs (95% confidence interval=0.1702–0.2058). These values approximate published values derived from sequence data
(23,
24), reflecting the high quality of the sequence data we obtained. Variants were observed throughout the mitochondrial genome but were significantly more frequent (F=3.25, df=21, 167, p=0.00001) in the control region (Tukey’s honest significant difference p≤0.05) and in NADH dehydrogenase 6 (Tukey’s honest significant difference p≤0.02), consistent with previous reports
(23,
24).
Case-Control Assays
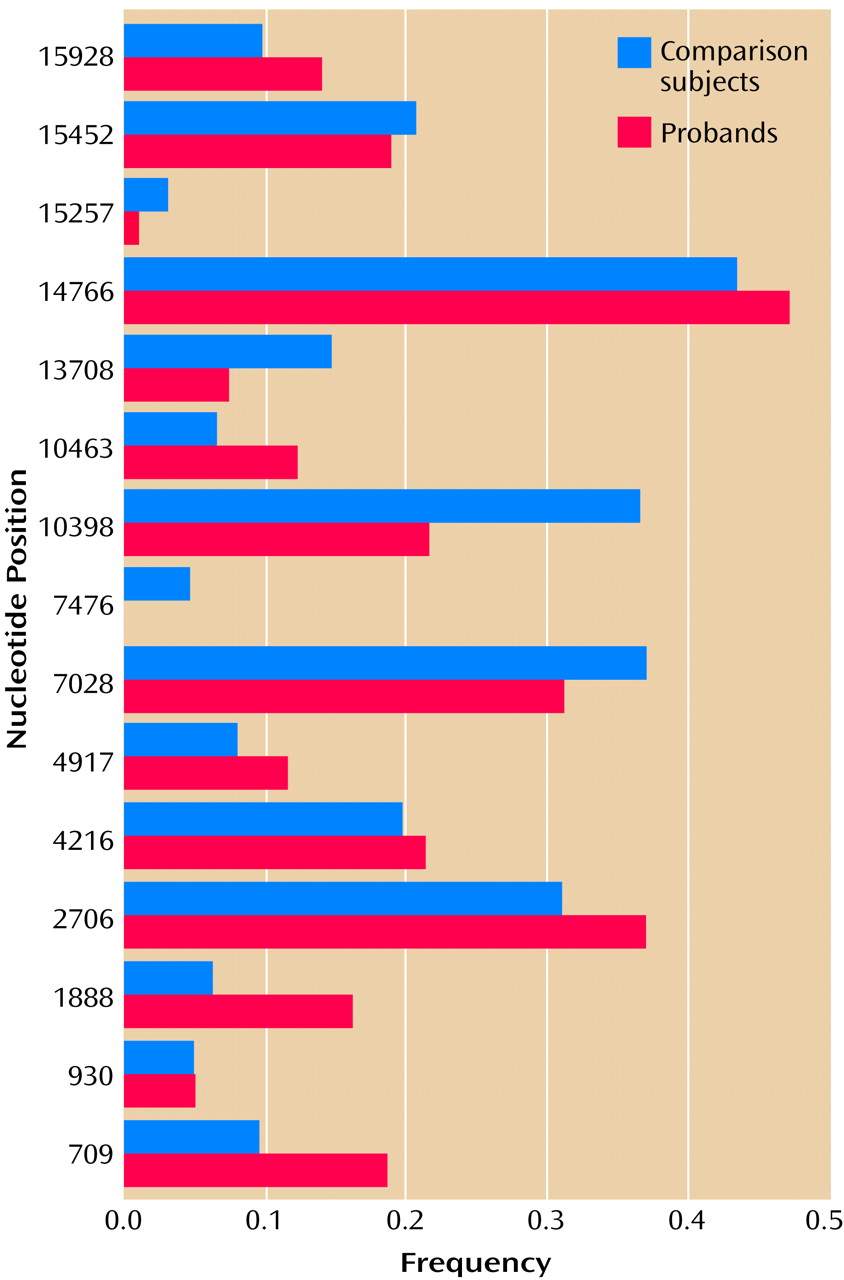
We selected 15 variants for further study in the case-control panel. They included all variants that were observed in more than one proband and that 1) resulted in an amino acid change, 2) occurred in genes encoding a tRNA or an rRNA, or 3) occurred outside of the hypervariable segments of the control region. The other variants were deemed unlikely to be of biological significance and were not pursued further.
The results of the case-control assay for 93 probands and 63 comparison subjects are shown in
Figure 3. There was no significant difference in the frequency of any variant in probands versus comparison subjects, although odds ratios of >2.0 or <0.5 were observed for four variants, at nucleotide positions 709, 1888, 10398, and 10463.
To rule out a potential role for these four variants in bipolar affective disorder, comparisons were repeated using probands and comparison subjects from the same mtDNA haplogroup. Two variants (nucleotide positions 1888 and 10463) were observed almost exclusively in subjects belonging to the T haplogroup, and almost all probands and comparison subjects within the T haplogroup carried these variants. Two variants (nucleotide positions 709 and 10398) were observed in subjects belonging to several different haplogroups. The rates of these variants in probands versus haplogroup-matched comparison subjects appeared quite similar, and the direction of any nominal differences between probands and comparison subjects varied across haplogroups.
Maternal Lineages
Because mtDNA is maternally inherited, variants that contribute to risk for bipolar affective disorder might be more common in probands who inherited the disorder through the maternal lineage. Thus we repeated all case-control comparisons after excluding the 23 probands from families in which bipolar affective disorder appeared to be paternally transmitted. The results were essentially unchanged (not shown). Again, there were no significant differences in the haplogroup distributions or in the frequencies of any variants in probands versus comparison subjects.
Discussion
This study was undertaken primarily to test the hypothesis that a point mutation in the mtDNA plays an etiologic role in bipolar affective disorder and contributes to the excess of maternal transmission previously observed in multiplex families
(4μ6). Complete mtDNA sequences were obtained from nine unrelated probands selected from extended pedigrees with a strict matrilineal transmission pattern. Of the 107 distinct variants detected by sequencing, 15 were assayed in a case-control panel to test for association with bipolar affective disorder. All of the major European haplogroups were represented at the expected frequencies in the probands and comparison subjects. Each variant was tested separately, and four variants showing nominal differences in the case-control analysis were tested again by using data for haplogroup-matched comparison subjects. All analyses were repeated after excluding probands from families with an apparently paternal transmission pattern. None of the variants we tested was associated with bipolar affective disorder in this sample. These results demonstrate that bipolar affective disorder occurs in each of the major groups of the European mtDNA haplotype structure and indicate that excess maternal transmission of bipolar affective disorder cannot be explained on the basis of point mutations in mtDNA.
This study is one of the first comprehensive investigations of the mitochondrial genome in bipolar affective disorder. Most previous studies have focused on bipolar affective disorder co-occurring with known mitochondrial diseases
(25–
28), with certain partial deletions of the mtDNA
(29,
30), or with abnormalities based on indirect measures of mitochondrial function
(31). Like one other published report
(32), the study reported here sought to characterize sequence diversity throughout the entire mitochondrial genome in subjects with bipolar affective disorder and to determine through direct sequencing whether any single variant or set of variants is etiologically important. We used a well-characterized sample of probands from families who had been systematically evaluated by psychiatrists and diagnosed by using standard research criteria. Because the apparent transmission pattern of illness within these families is generally known
(10), it was possible to select probands for sequencing from extended pedigrees with exclusive maternal transmission and to classify most probands in the case-control panel on the basis of an apparent maternal or nonmaternal transmission of bipolar affective disorder in their families
(4).
By sequencing mtDNA from nine probands, the study design was likely to detect all mtDNA variants present in at least 18% of the other 93 probands studied. Less common mtDNA variants—even those that might play an important etiologic role in some families—could have been missed. However, it is difficult to see how a variant present in less than 18% of probands could account for the exclusive maternal transmission of bipolar affective disorder observed in about 30% of families in this sample
(4).
The case-control panel we studied had good power to detect an association with a variant that increased the risk of bipolar affective disorder at least 2.5-fold, comparable to the increased risk of illness for the maternal relatives in this sample
(4). However, associations with variants conferring a smaller risk or with variants that increase risk only in conjunction with particular nuclear alleles or within a particular mtDNA haplogroup might not have been detected. Such associations would be difficult to detect without large samples of probands and comparison subjects within each subgroup. We did not attempt to detect deletions or rearrangements of the mtDNA that may have been present in this sample. Deletions and rearrangements are usually not preserved in DNA derived from lymphoblastoid cell lines, and mtDNA deletion syndromes are usually not transmitted maternally
(9).
This study of the mitochondrial genome in a psychiatric disorder illustrates some of the new issues that will arise for genetic studies of psychiatric disease once the entire sequence of the human genome is known and all genes have been identified. First, we observed considerable sequence diversity in the nine mitochondrial genomes we sequenced—approximately one variant in every 532 base pairs, comparable to previously reported values
(23,
24). Each mitochondrial genome sequenced in this study carried between 21 and 48 variants, compared to the Cambridge sequence. Comparable degrees of sequence diversity are beginning to be recognized in nuclear genes, similarly complicating attempts to associate particular variants with disease
(33,
34). Second, knowledge about the underlying haplotype structure of the mtDNA was helpful for ruling out potential differences in rates of particular variants that actually reflected chance differences in the haplogroup distributions in the probands and comparison subjects we studied. Nuclear gene association studies will also need to take background haplotype structure into account if false positive and false negative associations are to be minimized
(35). Third, we have demonstrated that probands with bipolar affective disorder occur in all of the major European mtDNA haplogroups. The occurrence of probands with bipolar affective disorder in all of these ancient haplogroup backgrounds suggests there has been no selection against particular mitochondrial lineages in bipolar affective disorder. This conclusion differs somewhat from that of Kirk et al.
(32), who found some evidence of selection against maternal lineages in probands with bipolar affective disorder. However, that study did not report haplogroup data, so the analyses may not be directly comparable. Because all of the subjects in the study by Kirk et al.
(32) resided in the same region of East Anglia, it is also possible that the population we studied was more diverse, making subtle selection effects difficult to detect.
Although our results argue against a role of the mitochondrial DNA in excess maternal transmission of bipolar affective disorder, further studies are needed to determine the true basis of this intriguing phenomenon. In any case, the search for major genes that determine the inherited susceptibility to bipolar affective disorder can now more confidently exclude the mitochondrial genome.