There is as yet no etiology-based treatment or cure for autism. Neurotransmitter signaling systems are relevant to symptom etiology and treatment and play a critical role in brain development. Investigations of autism have included only a few transmitters, including serotonin, dopamine, noradrenaline, and several neuropeptides
(1). These studies have mostly involved measurements in blood or cerebrospinal fluid and response to pharmaceutical agents.
To our knowledge, the cholinergic system has not previously been investigated neurochemically in autism, and yet there have been reports of pathological abnormalities in basal forebrain (septal) cholinergic neurons, such as larger than normal size and numbers in children and small size and numbers in adults
(2). Cholinergic afferents innervate the cerebral cortex during the most dynamic periods of neuronal differentiation and synapse formation, suggesting they play a regulatory role in these events
(3). In the rodent cortex, cholinergic innervation is the latest of the modulatory afferent innervations to mature
(4). In the human cerebral cortex and hippocampus, measures of cholinergic activity, including levels of choline acetyltransferase (the enzyme synthesizing the transmitter) and high-affinity nicotinic receptor binding, demonstrate substantial changes postnatally
(5,
6), during the early postnatal period, when symptoms of autism first become manifest
(7). Disruption of cholinergic innervation during early postnatal development (e.g., neonatal basal forebrain cholinergic lesions in rats) results in delayed cortical neuronal development and permanent changes in cortical cytoarchitecture and cognitive function
(3). Abnormalities in cortical cytomorphology, including abnormal thalamic afferent distribution in layer IV
(8–
10), are said to resemble pathologies associated with developmental disorders resulting in mental retardation
(11).
In view of the neuropathological evidence for involvement of the basal forebrain cholinergic system in autism, and the importance of this system in cortical development, cholinergic biomarkers were investigated in the basal forebrain and cerebral cortex (which receives a dense innervation from basal forebrain cholinergic neurons) in autistic individuals. Using available brain bank material, we analyzed tissue samples for the following biomarkers: 1) choline acetyltransferase biochemical activity, which is specific for cholinergic neurons; its decline is used as a standard measure of cholinergic neuronal loss or dysfunction
(12); 2) acetylcholinesterase activity as a relatively specific cholinergic marker; 3) cholinergic receptors located pre- and postsynaptically, including muscarinic M
1 and M
2 and high- and low-affinity nicotinic subtypes; and 4) both nerve growth factor and brain-derived neurotrophic factor, which control cholinergic neuronal function.
Method
Subjects
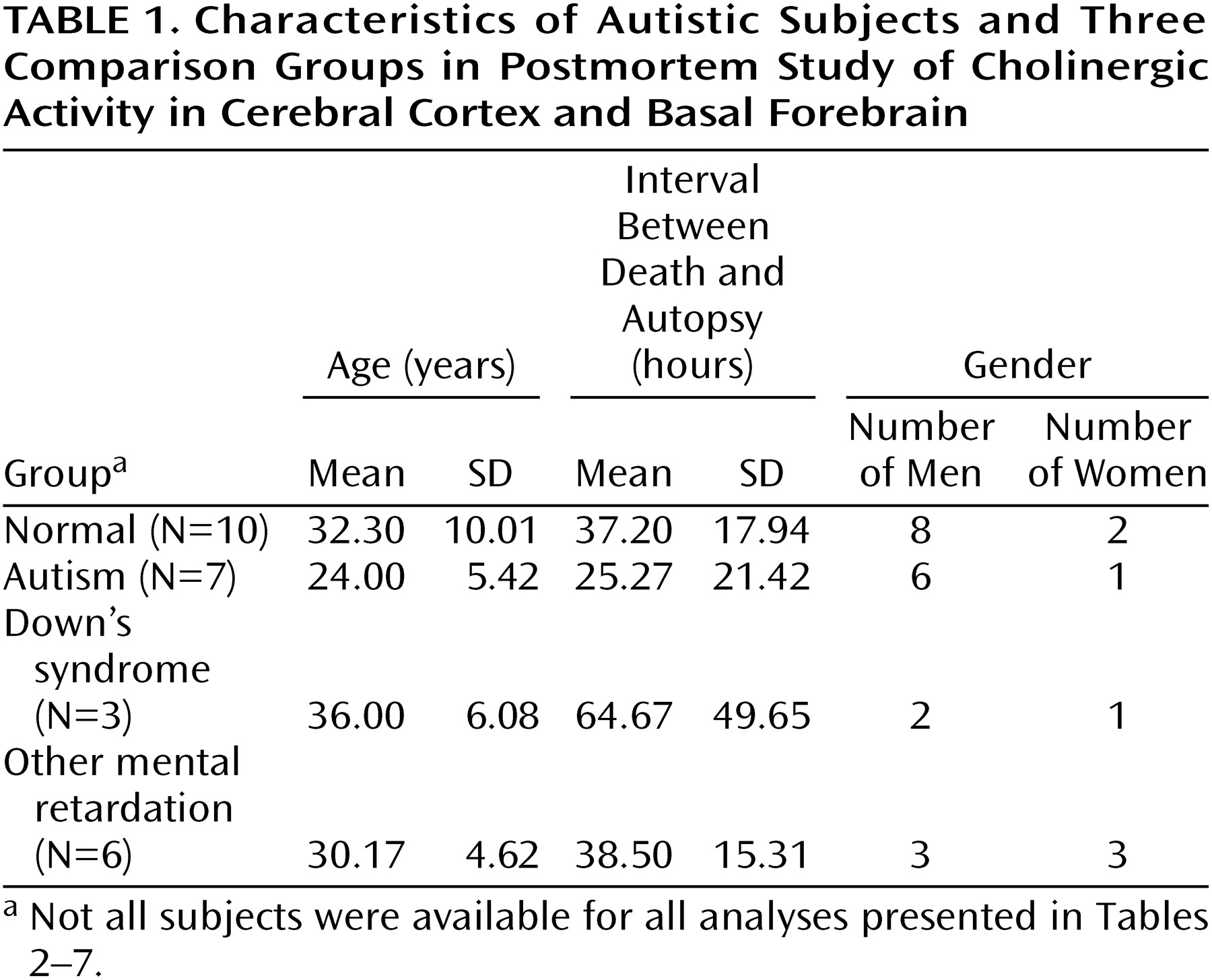
Autopsy tissue was examined from seven adults with autism meeting the DSM-IV criteria (which include delay or abnormal functioning in at least one of the following: social interaction, language, or symbolic or imaginative play). Clinical details were elicited from the parents by using the revised Autism Diagnostic Interview
(13). Ratings for the different interview areas were as follows: communication, nonverbal, 13–21; social, 22–25; behaviors, 5–10; and development, 2–5. For all subjects for whom information was available, there was clinical evidence of mental retardation (IQ, <20 to <50), and for 50% there was evidence of epilepsy. The causes of death for the autistic subjects were cardiac arrest, pneumonia, asphyxia, chronic renal failure, and fire. Comparisons were made with 10 normal individuals without mental retardation, with six individuals affected by other congenital cerebral disorders involving intellectual impairment (associated with birth trauma, viral encephalopathy, congenital cerebellar degeneration, or congenital epilepsy), and with three adults with Down’s syndrome. There was no history of mental retardation in the normal group, and the causes of death were respiratory failure (traffic accident), asphyxia (laryngeal injury), drug overdose, myocardial infarction or left ventricular failure, and pneumonia. Age, postmortem delay, and gender are provided in
Table 1. There were no significant differences between the groups in mean age or postmortem delay.
Frozen tissue was obtained from the Newcastle Brain Bank (United Kingdom), Harvard Brain Tissue Resource Center (United States), and University of Miami Brain and Tissue Bank for Developmental Disorders (United States); the samples for five of the seven autistic subjects were from the Harvard center. For each of the Harvard subjects, written consent approved by the McLean Hospital institutional review board was obtained from the legal next of kin, authorizing donation of tissue to the bank for use in research. For the Newcastle series, permission for postmortem study and for brain donation were obtained by prior consent from the next of kin, in accordance with the regulations of the North Tyneside Health Authorities Joint Ethics Committee.
For the Newcastle series, 1-cm coronal sections of the left hemisphere were snap frozen in liquid chlorodifluoromethane and stored at –70°C; the right hemisphere was fixed in paraformaldehyde. Tissue from the Harvard Brain Tissue Resource Center was subdissected at –20°C from whole hemispheres (three right and two left) originally frozen intact. The cortical tissue included the frontal cortex (Brodmann’s area 9) and parietal cortex (Brodmann’s area 39). The basal forebrain included the area just caudal to the anterior commissure and ventral to the globus pallidus, within the region of the substantia innominata.
Receptor Autoradiography
Frozen tissue blocks were subdissected at –20°C and mounted onto aluminum chucks. Then 10-μm cryostat sections were cut at –12°C and thaw mounted onto slides coated with Vectabond (Vector Laboratories, Burlingame, Calif.). The sections were air dried at room temperature for 2 hours before storage at –70°C. Adjacent sections were used to compare total with nonspecific binding; those determinations were made in duplicate and single, respectively.
Muscarinic M
1 receptor binding was measured by using 2 nM [
3H]pirenzepine with incubation for 1 hour; binding was assessed in the presence and absence of 1 μM atropine
(14). For M
2 receptor binding, 7 nM [
3H]AFDX-384 (2,3-dipropylamine) was used with displacement by 1 μM atropine and incubation for 1 hour
(15). For epibatidine binding to the nicotinic receptor (α
3/β
2 or α
4/β
2 subunit), sections were incubated with 1 nM (±)[
3H]epibatidine for 3 hours at room temperature, and nonspecific binding was assessed in the presence of 1 μM cytisine
(6). [
125I]α-Bungarotoxin binding (α
7 subunit) was measured by using 1.2 nM radioligand, and nonspecific binding was measured in the presence of 2.5 mM nicotine during both the pre-incubation period (30 minutes) and incubation (2 hours)
(6). The sections were washed and dried after incubation and exposed to high-performance autoradiography film with high and low tritium standards for 3 weeks for pirenzepine, 2 months for AFDX-384, 3 months for epibatidine, and 1 week for α-bungarotoxin. After development, the images were quantified by using a Lynx densitometry system (Applied Imaging International, Newcastle Upon Tyne, U.K.).
Membrane Receptor Binding
For nicotine binding to the basal forebrain, membranes were prepared
(16) and incubated in triplicate with 2 nM [
3H]nicotine with or without unlabeled 1 mM nicotine. The membranes were collected on Whatman GF/C glass fiber filters (presoaked in 0.3% polyethyleneimine) by using a PHD cell harvester (model 2000, Cambridge Technology, Watertown, Mass.). The filters were washed three times in buffer, and the beta radiation was counted by using a scintillation counter.
For the determination of the concentrations producing 50% inhibition (IC
50) of epibatidine and pirenzepine binding, washed membrane pellets were prepared from subjects for whom sufficient tissue from the parietal cortex was available (three normal subjects and two autistic subjects). Protein was measured by the method of Lowry et al.
(17). For epibatidine binding, resuspended membranes were pre-incubated with 1 × 10
–12 to 10
–8 M unlabeled epibatidine, before the addition of 1 nM [
3H]epibatidine, and incubated for 3 hours at room temperature. For the M
1 receptor, 1 × 10
–11 to 10
–4 M unlabeled pirenzepine and 2 nM [
3H]pirenzepine were used with 1 hour of incubation. The membrane suspensions were filtered through Whatman GF/C filters (presoaked in 0.5% polyethyleneimine), and beta radiation was determined by using a liquid scintillation counter. Binding was analyzed by using nonlinear regression and assuming a one-site competition model (GraphPad Prism, GraphPad Software, San Diego).
Nicotinic Receptor Subunits
In select samples of the parietal cortex, nicotinic receptor subunits α
3, α
4, α
7, and β
2 were quantified by using Western blotting
(18). The polyclonal antibodies used were as follows: antihuman nicotinic acetylcholine receptor α
3 antibody (antibody 607), against the carboxy terminus of the nicotinic acetylcholine receptor
(19); sc-1772 raised against the carboxy terminus of the nicotinic acetylcholine receptor α
4 subunit (Santa Cruz Biotechnology, Santa Cruz, Calif.); sc-1447, against the carboxy terminus of the α
7 subunit (Santa Cruz Biotechnology); and antihuman nicotinic acetylcholine receptor β
2 subunit raised in rabbit against a human epitope, corresponding to an amino acid sequence in the cytoplasmic loop region at position 350–452 between the M
3 and M
4 transmembrane domains
(20). Membrane pellets, prepared as already described, were subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis and electroblotting onto polyvinylidene fluoride membranes as described previously
(18). The samples were evaluated by Western blotting between three and five times.
Choline Acetyltransferase
Representative samples of gray matter (including all cortical layers) were removed, and 10% homogenates of these and basal forebrain samples were prepared in 0.32 M sucrose containing 0.5% Triton X-100 at 4°C. Duplicate or triplicate samples were incubated at 37°C for 20 minutes with [
14C]acetyl coenzyme A by using, for the cortical samples, an adaptation
(21) of the method of Fonnum
(22) and, for the basal forebrain samples, the original Fonnum method
(22).
Acetylcholinesterase
For histochemical analyses of the cortical samples, 10-μm cryostat sections were fixed in formal calcium and incubated with acetylthiocholine iodide with staining intensification as previously described
(23). Acetylcholinesterase biochemical activity in basal forebrain homogenates was measured in duplicate according to the method of Ellman et al.
(24) in the presence of 1 × 10
–5 M iso-octamethyldiphosphoramide, a specific inhibitor of butyrylcholinesterase activity.
Nerve Growth Factor and Brain-Derived Neurotrophic Factor
Two-site enzyme immunoassays were performed to measure endogenous levels of brain-derived neurotrophic factor and nerve growth factor. Forebrain samples were sonicated in 50 mM Tris HCl, pH 7.0, 150 mM NaCl, 1% bovine serum albumin, 1% Triton X-100, 4 μg/ml aprotinin and 0.5 mM phenylmethylsulfonyl fluoride, 2 mM EDTA, 0.1 mM benzethonium chloride, and 0.05% sodium azide, and they were then centrifuged (15,000
g) for 10 minutes. Nerve growth factor and brain-derived neurotrophic factor standards (1 to 500 pg/ml) were used to generate standard curves. The immunoplates were coated for 2 hours with purified monoclonal antibodies to nerve growth factor (0.67 μg/ml) and brain-derived neurotrophic factor (100 ng/well), and tissue extracts (in duplicate with 10 mM CaCl
2) were added. The trophic factors were quantified according to modifications of previously published methods
(25,
26).
Radiochemicals
[3H]Pirenzepine (85.6 Ci/mmol), [3H]AFDX-384 (106.5 Ci/mmol), (±)[3H]epibatidine (33.8 Ci/mmol), (3-[125I]iodotyrosyl)-α-bungarotoxin (150 Ci/mmol), [3H]nicotine (81.5 Ci/mmol), and [14C]acetyl coenzyme A (58.9 mCi/mmol) were purchased from New England Nuclear Life Science Products, Inc. (Boston).
Statistical Analysis
Since the different brain areas are functionally distinct, they were analyzed separately. Values for cortical nicotinic and muscarinic binding were analyzed by two-way analysis of variance (ANOVA) (general linear model, Minitab release 12 [Minitab, State College, Pa.]) with group and cortical layer as factors. If a statistically significant effect was demonstrated for group and/or cortical layer but there was no significant interaction between the factors, the data were analyzed as suggested by Kinnear and Gray
(27), by one-way ANOVA followed by post hoc analysis using Fisher’s pairwise comparison, with significance set at p<0.05 (Minitab release 12). Values for the forebrain were analyzed by t tests.
Results
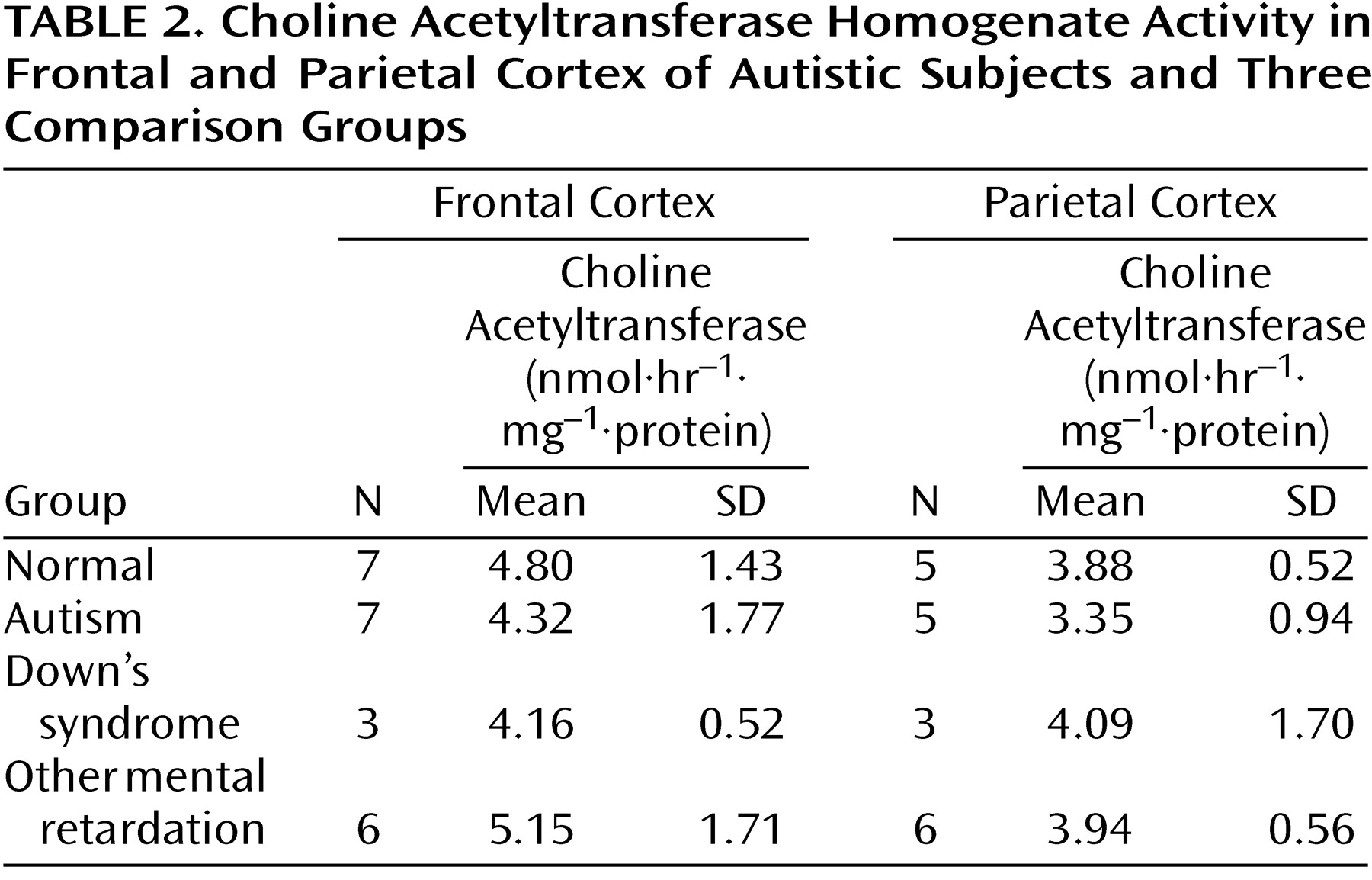
There were no significant abnormalities in the autistic or nonautistic groups of the presynaptic marker, choline acetyltransferase, in the frontal cortex, parietal cortex, or basal forebrain (
Table 2 and
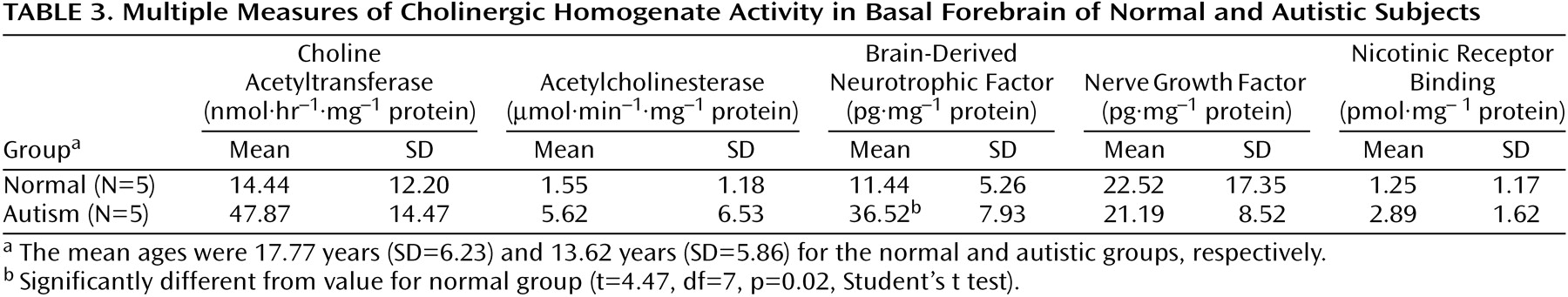
Table 3), although there was a nonsignificant tendency for the activity to be higher in the basal forebrain in the autistic group. Since the values for cortical choline acetyltransferase activity did not differ significantly among groups, a detailed analysis of the acetylcholinesterase histochemical reactivity in the cortex was not undertaken. It was established by semiquantitative macroscopic assessment that the histochemical activity levels were similar in the autistic and normal groups. In the basal forebrain there was also no significant difference in the acetylcholinesterase activity measured biochemically, although, as with choline acetyltransferase, there was a nonsignificant tendency toward greater activity in the autistic group (
Table 3). Of the two neurotrophin measurements investigated in the basal forebrain, the levels of nerve growth factor did not differ significantly, but the levels of brain-derived neurotrophic factor were significantly higher in the autistic group, three times as high as in the normal comparison group (
Table 3).
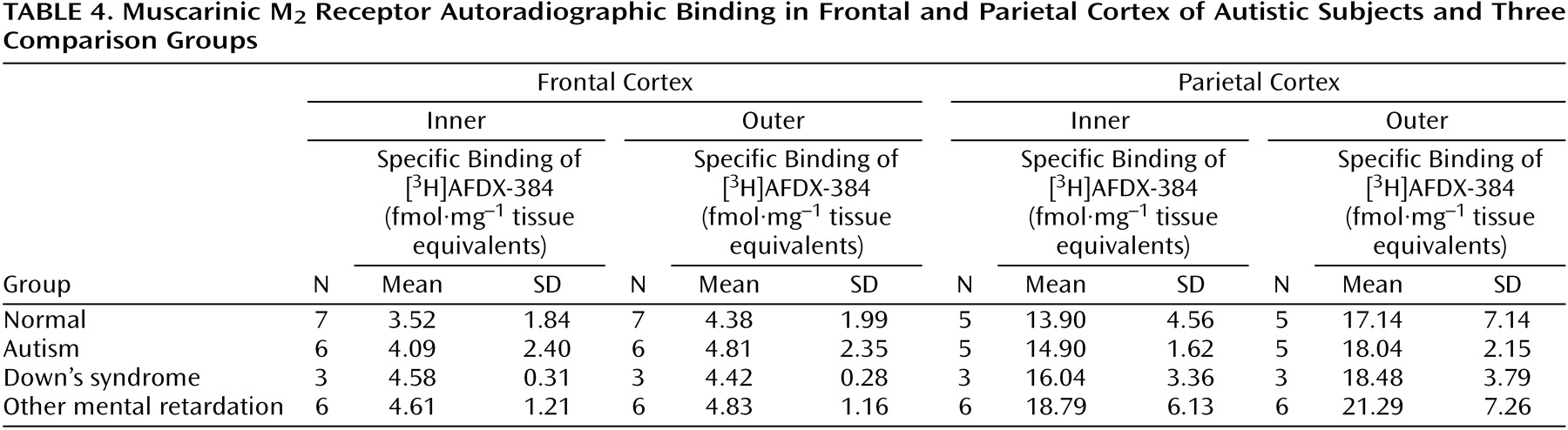
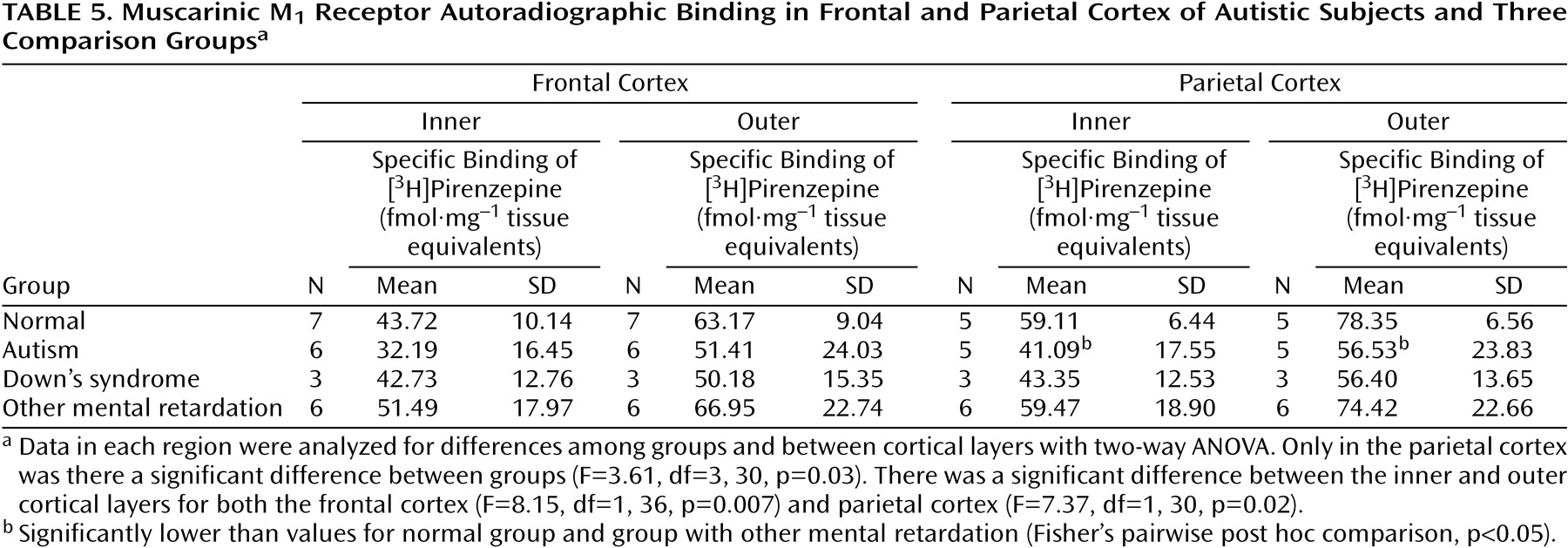
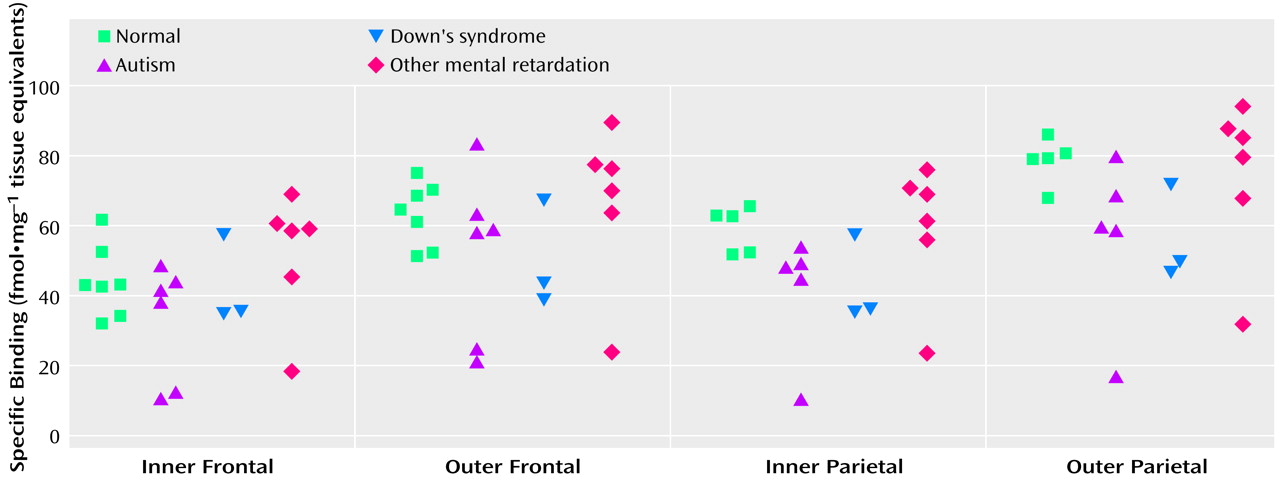
Binding to the M
2 receptor in the two cortical areas did not differ substantially between the autistic and other groups (
Table 4). In contrast, cortical M
1 receptor binding was lower in the autistic group (
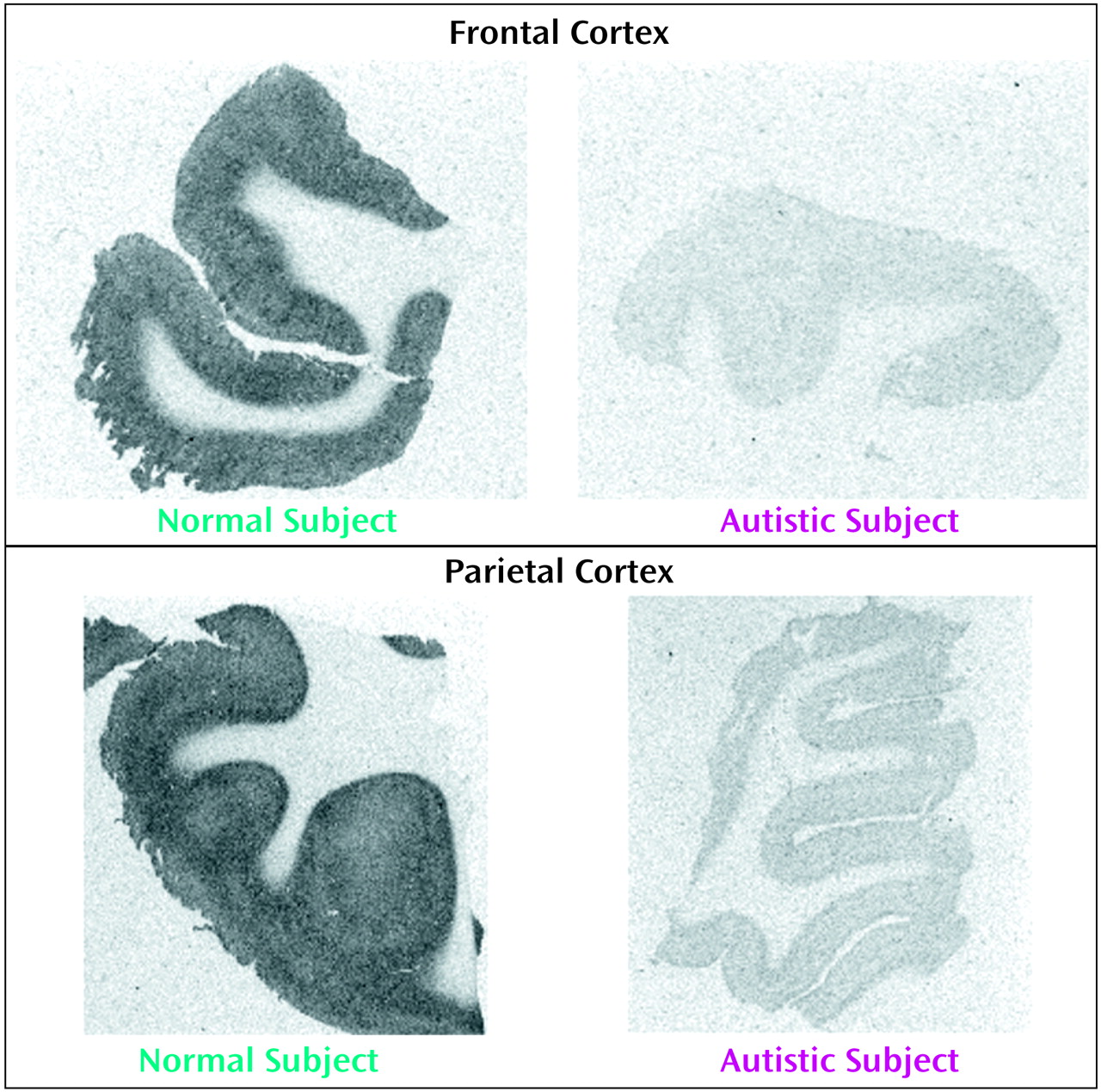
Table 5,
Figure 1,
Figure 2>). This difference was apparent in both cortical areas and in both the outer and inner cortical layers, and for the parietal cortex the autistic group was significantly different from both the normal group and the group with other mental retardation. There was, with the exception of one case, no overlap in M
1> receptor binding in the inner parietal cortex between the autistic and normal group or group with other mental retardation. The IC
50 values for pirenzepine displacement were 1.86×10
–9 and 2.03×10
–9 M in the two autistic subjects, whereas the mean for the three normal subjects was 1.68×10
–9 M (SD=0.28×10
–9).
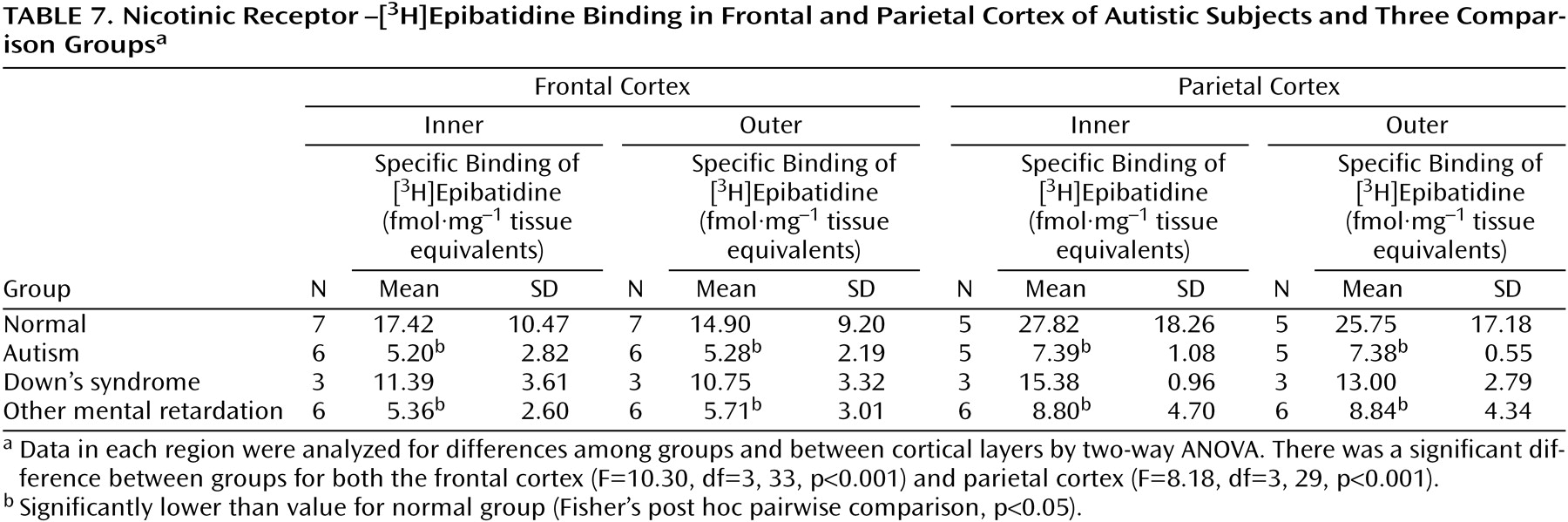
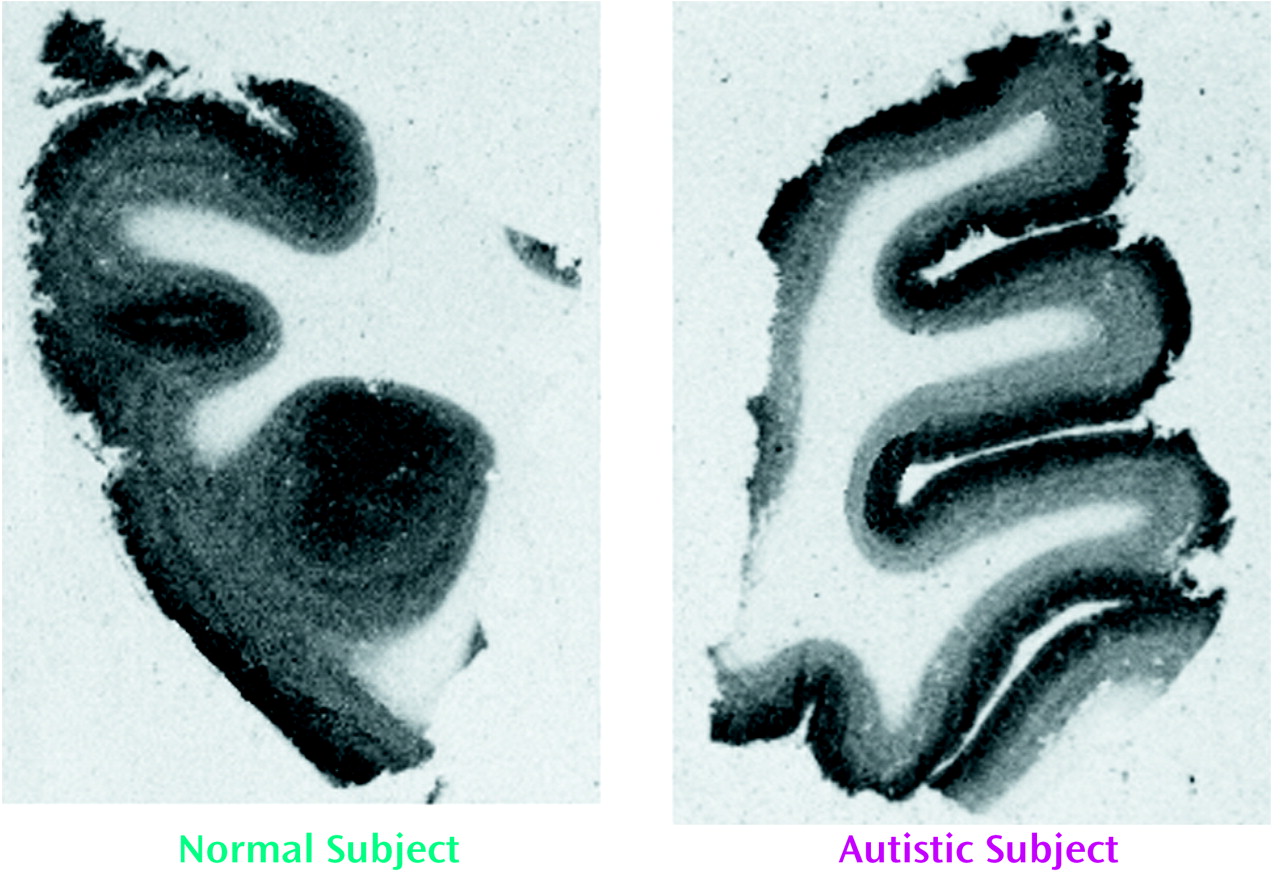
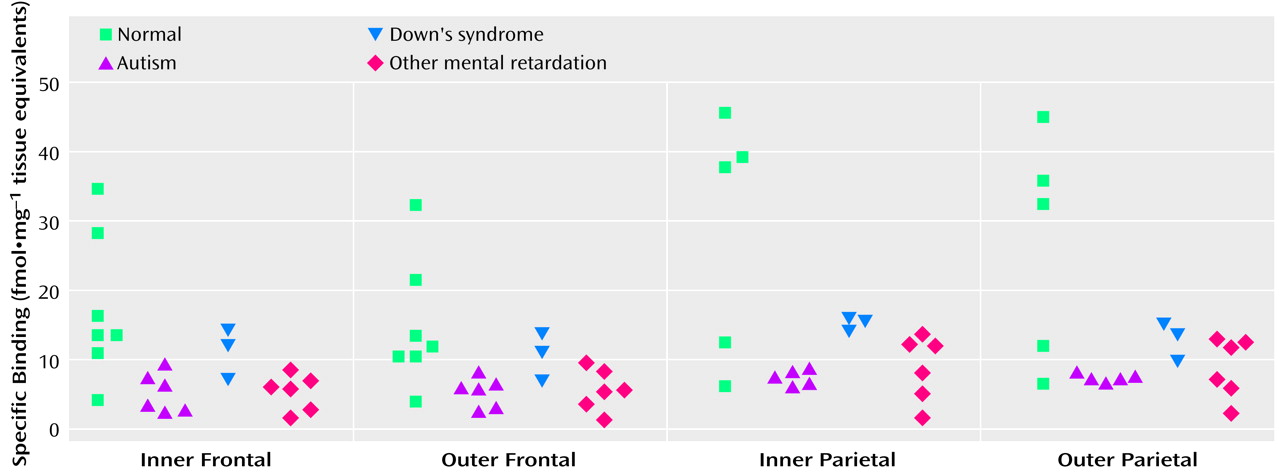
With respect to nicotinic receptor binding, there was no abnormality of α-bungarotoxin binding in either cortical area in the autistic group (
Table 6). By contrast, in both cortical areas the autistic group had significantly and substantially lower epibatidine binding (to 27%–35% of normal) than was found for the normal comparison group (
Table 7,
Figure 3,
Figure 4); the differences were apparent in both the outer and inner cortical layers. Epibatidine binding was also significantly lower in the nonautistic mental retardation group, but not in the Down’s syndrome group, than in the normal group. It is unlikely that the lower receptor level is due to a difference in tobacco use between the groups. Although smoking histories were not available for the series, if 50% of the normal group smoked (a rate higher than for the general population) and none of the autistic subjects smoked, the expected difference in epibatidine binding—based on a smoking-induced doubling of binding in the normal human cortex
(28)—would not exceed 35%. Moreover, in the basal forebrain, [
3H]nicotine binding was normal (
Table 3). IC
50 values for epibatidine binding in the parietal cortex were 7.9×10
–10 M and 2.3×10
–10 M for the two normal subjects and 9.1×10
–10 M and 10.7×10
–10 M for the two autistic subjects. According to Western blotting of samples from the parietal cortex, there was lower α
4 and β
2, but not α
3 or α
7, immunoreactivity in autism. The α
4 immunoreactivity was in two cases 2.2% and 5.1% of the mean value for the normal group, and β
2 immunoreactivity was 18.1% and 43.1%.
There was no difference in cortical epibatidine or pirenzepine binding between normal subjects whose samples had been slow or snap frozen. The mean values for the inner frontal cortex for five snap-frozen and two slow-frozen samples were, respectively, 16.1 and 21.0 fmol·mg–1 for epibatidine binding and 39.9 and 42.7 fmol·mg–1 for pirenzepine binding.
Discussion
This study indicates the potential importance of specific cholinergic receptors and neurotrophins in developmental neurobiology. The findings indicate that although cholinergic enzyme markers are normal in autism, many nicotinic (high-affinity) and moderate muscarinic M1 receptor measures in the cerebral cortex are lower than normal, and the level of brain-derived neurotrophic factor in the basal forebrain is higher than normal.
Normal Presynaptic Cholinergic Activity
In relation to the original hypothesis of basal forebrain cholinergic dysfunction, the finding of normal choline acetyltransferase activity both in the frontal and parietal cortex and in the basal forebrain suggests that the presynaptic cholinergic innervation of the cortex is structurally intact in autistic subjects. This indicates either that cholinergic neurons are not depleted or that compensatory axonal sprouting has occurred. In another developmental disorder, Rett’s syndrome, in which levels of choline acetyltransferase and the vesicular acetylcholine transporter are lower than normal in various areas, including the cortex
(29,
30), disruption of cholinergic innervation is likely due to developmental or degenerative neuronal abnormalities occurring before or shortly after birth in the absence of compensation.
Low Binding of Muscarinic M1 Receptors
The low M
1 receptor binding apparent in the parietal cortex of autistic subjects but not subjects with other types of mental retardation indicates a specific abnormality in cholinoceptive function, since the M
1 receptor is located postsynaptically. The binding abnormality reflected a low number of receptors. This could be related to epilepsy, which occurs in up to 40% of autistic children
(31), since a low number has been reported in hippocampal sclerosis associated with temporal lobe epilepsy
(32). The finding that a similar series of subjects had normal pirenzepine binding in the hippocampus
(33), an area particularly susceptible in epilepsy, suggests another basis for the low number of receptors, such as dendritic dysfunction.
Low Binding of High-Affinity Nicotinic Receptors
The lower degree of high-affinity nicotinic receptor binding in the cortex of the autistic subjects was extensive and likely, on the basis of the kinetic and immunochemical evidence, to reflect low receptor numbers. The lower epibatidine but not α-bungarotoxin binding is consistent with the preliminary immunoblot findings of lower immunoreactivity of the nicotinic α
4 and β
2 subunits. Although values for the α
4 subunit and high-affinity nicotinic agonist binding site are elevated as a result of exposure to nicotine
(18,
34), the lower levels of receptor binding in the autistic individuals in this study could not be attributed to differences in tobacco use between the different groups.
Relationships between low nicotinic receptor binding and synaptophysin identified in another cerebral disorder, Alzheimer’s disease
(35), suggest that the low receptor binding in autism may be associated with abnormal cortical neuronal morphology, possibly involving overextensive synaptic pruning. Knockout (β
2) mice lacking nicotinic receptor binding develop a degree of age-related cortical atrophy and neuronal loss in conjunction with cognitive impairment
(36). Lower binding at the high-affinity nicotinic receptor agonist site not only in autism but also in the nonautistic mental retardation group could indicate that this condition is not specific to autism and that receptor loss may be a consequence rather than cause of cortical dysfunction. However, developmental brain abnormalities occur in both groups and an overlap in the processes involved would be expected.
Nicotinic agents are analgesic
(37), and on the basis of a gene knockout model
(38) the α
4 subunit has been implicated in pain perception. Low levels of this receptor subtype in autism may thus be associated with the low degree of pain reactivity that is present in the disease
(39). Since abnormal galvanic skin responses (peak amplitudes three times normal, number of arousal events per minute two times normal, and absence of a baseline) have been observed in autism (unpublished 2000 study of P. Iversen and H. Ramshandran), and since such responses may depend on the integrity of sympathetic cholinergic neural pathways
(40), it may be worth investigating the extent to which the central nervous system cholinergic abnormalities reported here extend to the peripheral nervous system. The finding that, in contrast to epibatidine binding, α-bungarotoxin binding is normal in autism is interesting since the gene coding for the α
7 subunit is located close to the q11-15 region of chromosome 15
(41), which is considered to be one of the likely sites of genetic abnormalities in autism
(42).
High Level of Brain-Derived Neurotrophic Factor
The finding that the level of brain-derived neurotrophic factor was significantly higher than normal in the basal forebrain can be interpreted in various ways. Since brain-derived neurotrophic factor is up-regulated by cholinergic activity in developing rat hippocampus
(43), it is unlikely that the abnormality is due to developmental cholinergic dysfunction. One explanation for the high level, since neurotrophins influence the development and function of basal forebrain cholinergic neurons
(3,
44), is that it reflects a regional compensatory mechanism. This may underlie the finding that levels of cholinergic biomarkers, such as choline acetyltransferase in the cortex and basal forebrain, were normal or even high. An alternative explanation is that overexpression of brain-derived neurotrophic factor is an intrinsic component of the autism disease process. The recent finding
(45) of high blood levels of brain-derived neurotrophic factor, among three brain peptides or proteins (brain-derived neurotrophic factor, vasoactive intestinal peptide, and calcitonin gene-related peptide), in 62 of 64 newborn autistic and other mentally retarded individuals suggests an intrinsic rather than compensatory mechanism.
Comparison With Schizophrenia
The overall pattern of cholinergic abnormalities in autism—low levels of muscarinic and nicotinic receptors in conjunction with normal levels of presynaptic cholinergic markers in the cortex—is more similar to that seen in schizophrenia than to patterns in other disorders involving a cholinergic abnormality, such as Rett’s syndrome, Alzheimer’s or Parkinson’s disease, head injury, or vascular dementia. There is in common with schizophrenia a sparing of the presynaptic marker, choline acetyltransferase, but low levels of muscarinic and nicotinic receptors, although in schizophrenia the α
7 subtype may also be involved
(46,
47). In schizophrenia there is also a high level of brain-derived neurotrophic factor but not nerve growth factor in cortical areas
(48). There is an extensive overlap in clinical symptoms between autism and schizophrenia, both behaviorally and cognitively, e.g., in conceptual abnormalities
(49). The same neural systems are likely to be involved in both, although differing in developmental staging and etiology.
Conclusions
If the low level of cortical nicotinic receptors is consistently observed and clinically relevant, therapeutic strategies could include receptor agonists, such as nicotine, which has already been applied in Tourette’s disorder with amelioration of symptoms
(50). Such treatment could also be disease modifying. Nicotine significantly increases branching of both axons and dendrites in cultured hippocampal neurons
(51). In adolescent rat brain, nicotine exposure results in persistent up-regulation of nicotinic receptors in a variety of brain areas, including the cortex
(52), and in adult mouse brain chronic nicotine exposure promotes retention of nicotinic receptors that decline in aging
(53).
On the basis of the present findings, the role of the cholinergic system in autism should be investigated further, with respect to other brain regions, additional cholinergic receptor subtypes, and clinical correlates and in younger individuals.