Recent investigations in schizophrenia have targeted glutamatergic neurotransmission in the thalamus because of both its central role in sensory processing and its rich glutamatergic connections to limbic regions of the brain
(1). Several lines of evidence have implicated thalamic abnormalities in subjects with schizophrenia. Studies that used magnetic resonance imaging or unbiased volume estimation techniques have reported thalamic volume deficits in patients with schizophrenia
(2–
7). Other work that used positron emission tomography or single photon emission computed tomography detected abnormalities in thalamic metabolism, while other studies have reported deficits in the number of thalamic neurons and glia in patients with schizophrenia
(5,
8–
14). The reported deficits in thalamic volume, metabolism, and cell number in patients with schizophrenia are consistent with other work that has demonstrated that persons with schizophrenia have deficits in recalling complex narrative material, which suggests an abnormality in cortico-thalamic-cerebellar connectivity
(11,
12). While these data suggest the thalamus as a candidate region for pathophysiology in schizophrenia, they do not indicate which neurochemical substrates are associated with these abnormalities. The glutamatergic system is a likely candidate, since pharmacological evidence has implicated glutamatergic dysfunction in schizophrenia.
Glutamate receptors are implicated in the pathophysiology of schizophrenia in part because phencyclidine (PCP) induces schizophreniform psychosis
(15–
19). PCP binds to the intrachannel site of the
N-methyl-
d-aspartate (NMDA) receptor, blocking normal receptor activity
(20,
21). Thus, abnormalities in thalamic glutamatergic neurotransmission may contribute to the pathophysiology of schizophrenia. Previous work in our laboratory has detected lower expression of some NMDA receptor subunits and binding sites in the thalamus of subjects with schizophrenia
(22). Striking alterations in AMPA and kainate subunit mRNA and binding site expression were not detected
(22), and there were no significant changes in the expression of transcripts encoding the metabotropic glutamate receptors in the thalamus in schizophrenia
(23). These results are consistent with the hypothesis that NMDA receptor hypoactivity may contribute to psychopathology in schizophrenia. In addition to the NMDA receptor, however, other synaptic elements, such as excitatory amino acid transporters (EAATs), may directly affect glutamatergic neurotransmission in schizophrenia.
Recently, a novel family of excitatory amino acid transporters was cloned and characterized
(24). This family of sodium-dependent glutamate/aspartate transporters includes five members (EAAT1–EAAT5) and is structurally similar to the neutral amino acid transporters ASCT1 and ASCT2 but is unrelated to other neurotransmitter transporters
(25,
26). Structural studies have predicted that these transporters possess six to 10 transmembrane domains, which may form an aqueous transmembrane pore
(27). The transporter is reversible when the sodium gradient is altered, such as during a seizure
(28). It is of interest that EAAT4 and EAAT5 appear to have the unique property of gating chloride ions
(29,
30). The transcripts have specific patterns of cellular localization: EAAT1 and EAAT2 have been localized to astroglia, whereas EAAT3 and EAAT4 are localized to neurons
(31–
37).
The best studied of the glutamate transporters, EAAT2 (called GLT-1 in the rodent), accounts for approximately 90% of rodent forebrain glutamate reuptake
(38,
39). Expression of EAAT2 protein and mRNA has been observed throughout the human brain but is highest in the forebrain
(31,
32). Astrocytes have been identified as the predominant cell type expressing EAAT2 protein by immunohistochemical staining
(33–
35). GLT-1 knockout mice exhibit hippocampal neurodegeneration and develop lethal seizures, emphasizing the physiologic importance of homeostatic regulation of glutamate levels
(38,
39).
Similar to EAAT2, EAAT3 (called EAAC1 in the rodent) protein expression in the human brain is detected in the frontal cortex and the hippocampus
(31). Postmortem human EAAT3 mRNA localization has not been evaluated. EAAT3 is localized to both post- and presynaptic neuronal soma and contributes approximately 40% of hippocampal glutamate transport
(34). In contrast, levels of EAAT1 (called GLAST in the rodent) and EAAT4 protein expression in rodent CNS are highest in the cerebellum, in the Bergmann glia and Purkinje cell types, respectively
(31,
36,
37). Human studies of EAAT1 protein expression indicate high levels in the frontal cortex, while EAAT1 and EAAT4 mRNA levels have not been extensively examined in humans. CNS EAAT5 mRNA expression is limited to the retina
(29).
Subtle functional differences between the glutamate transporters illustrate the complexity of the glutamate synapse. For example, EAAT2, but not EAAT1, is inhibited by dihydrokainate, suggesting that anatomic or circuit-dependent differences in glutamate transporter pharmacology may reflect differences in the functional nature of a given glutamate synapse or circuit
(40,
41). The variability in effects of transporter antisense or knockout experiments is consistent with this notion. GLT-1 (EAAT2) knockout animals develop lethal seizures, while GLAST (EAAT1) knockout animals have subtler defects, including difficulties with coordination
(38,
39,
42). It is not surprising that alterations in excitatory amino acid transporter expression have been reported in schizophrenia, Huntington’s disease, and amyotrophic lateral sclerosis
(43–
48). It is interesting to note that variable splicing of EAAT2 in amyotrophic lateral sclerosis subjects has been detected, implicating alterations in mRNA processing as a possible etiology for abnormalities in excitatory amino acid transporter expression in human diseases
(45). Thus, we hypothesized that in schizophrenia, an abnormality in excitatory amino acid transporter expression may contribute to glutamatergic dysfunction. The aim of this study was to investigate thalamic expression of members of the family of excitatory amino acid transporters (EAAT1, EAAT2, EAAT3, and EAAT4) in the brain of subjects with schizophrenia.
Method
Subjects
Twelve subjects with schizophrenia (five women and seven men; mean age=70 years [SD=8], mean postmortem interval=420 minutes [SD=328]) and eight individuals with no psychiatric illness (six women and two men; mean age=77 years [SD=14], mean postmortem interval=613 minutes [SD=425]) from the Mount Sinai Medical Center Brain Bank were studied. The schizophrenic subjects in the present study are the same as those used in our previous reports on glutamate receptor expression in the thalamus in schizophrenia
(22,
23). Subjects were classified as having schizophrenia if 1) the presence of schizophrenic symptoms could be documented before age 40; 2) the medical records contained evidence of psychotic symptoms and at least 10 years of psychiatric hospitalization with a diagnosis of schizophrenia; 3) the DSM-III-R diagnosis of schizophrenia was agreed upon by two experienced clinicians; and 4) neuropathological examination did not reveal Alzheimer’s disease or other degenerative disorders. Subjects with a history of alcoholism or substance abuse were excluded from this cohort. Neither age (t=1.6, df=18, p>0.10), postmortem interval (t=1.1, df=18, p=0.30), nor sex distribution (χ
2=2.15, df=1, p>0.10) were significantly different between the two groups. At the time of death of the persons with schizophrenia, six were receiving antipsychotics, five had a mean drug-free period of 5.8 weeks (SD=3.4), and one subject had been drug free for 416 weeks.
In Situ Hybridization
Brains were obtained at autopsy, and 20-μm sections were prepared as previously described
(22,
23). Expression of EAAT1, EAAT2, EAAT3, and EAAT4 mRNA was determined by using in situ hybridization. To generate subclones from which to synthesize riboprobes, we amplified unique regions of EAAT1 (National Center for Biotechnology GenBank assession number: U03504, nucleotide coding region: 526–825), EAAT2 (NM004171, 601–1026), EAAT3 (NM004170, 156–979), and EAAT4 (NM005071, 541–900) from a human cDNA brain library (EdgeBiosystems, Gaithersburg, Md.) by using polymerase chain reaction (PCR). For EAAT2, the region selected binds to all known splice variants. Amplified cDNA segments were extracted (QIAquick Gel Extraction Kit, Qiagen, Valencia, Calif.), subcloned (Zero Blunt TOPO PCR cloning kit, Invitrogen, Carlsbad, Calif.), and confirmed by nucleotide sequencing (Thermo Sequenase Radiolabeled Termination Cycle Sequencing Kit, USB, Cleveland). Riboprobes were synthesized from linearized plasmid DNA, and in situ hybridization was performed as previously described
(22). Briefly, radiolabeled probes were diluted in 50% (vol:vol) formamide hybridization buffer, applied to sections, and incubated overnight at 55°C. In situ hybridization was performed with sense and antisense probes for EAAT1, EAAT2, EAAT3, and EAAT4 on serial sections of macaque thalamus to confirm probe specificity.
Data Analysis
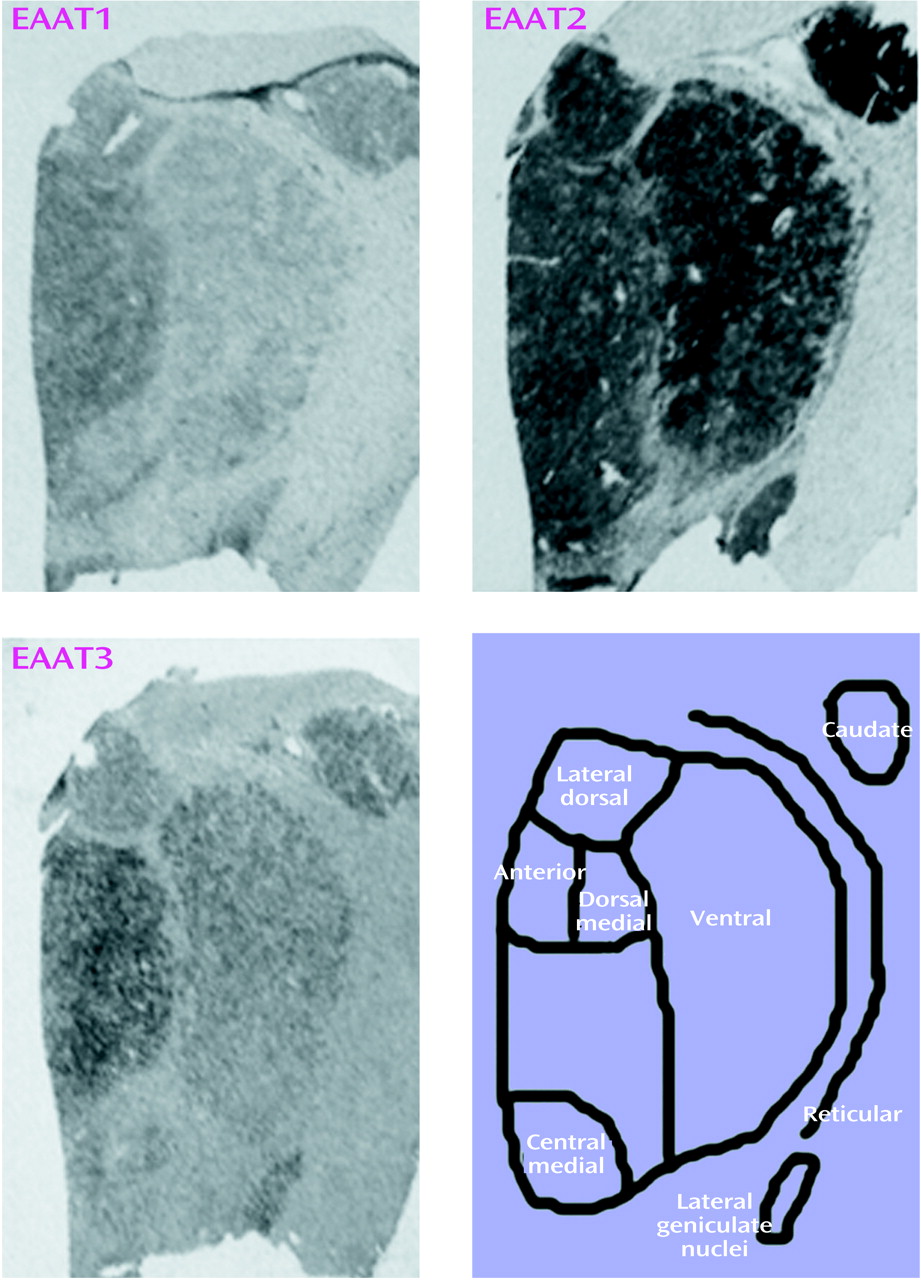
Thalamic nuclei were identified in each section on the basis of cellular and white matter patterns, defined by cresyl violet and gold chloride staining of adjacent sections from each subject as previously described
(22,
23). Subdivisions of thalamic nuclei were not clearly distinguishable in all sections for all subjects, and subdivisions were pooled for data analysis. The following nuclei were identified for each subject: anterior, dorsal medial, lateral dorsal, central medial, ventral, and reticular. In this study, “ventral” nucleus corresponds to a grouping of several nuclei of the ventral tier. Images were digitized from films and analyzed with Image 1.56 (National Institutes of Health, Bethesda, Md.). For digitized images, gray-scale values of tissue background were subtracted from values for each nucleus and converted to optical densities. Values from two sections for each subject were averaged and used for statistical analysis. Statistical analysis was performed for each probe by two-way analysis of variance, with diagnosis and nuclei as the independent variables. Post hoc analyses were by the Newman-Keuls test. We analyzed the relationship between age and excitatory amino acid transporter expression in each nucleus with a series of correlation coefficients. For all tests, alpha=0.05.
Results
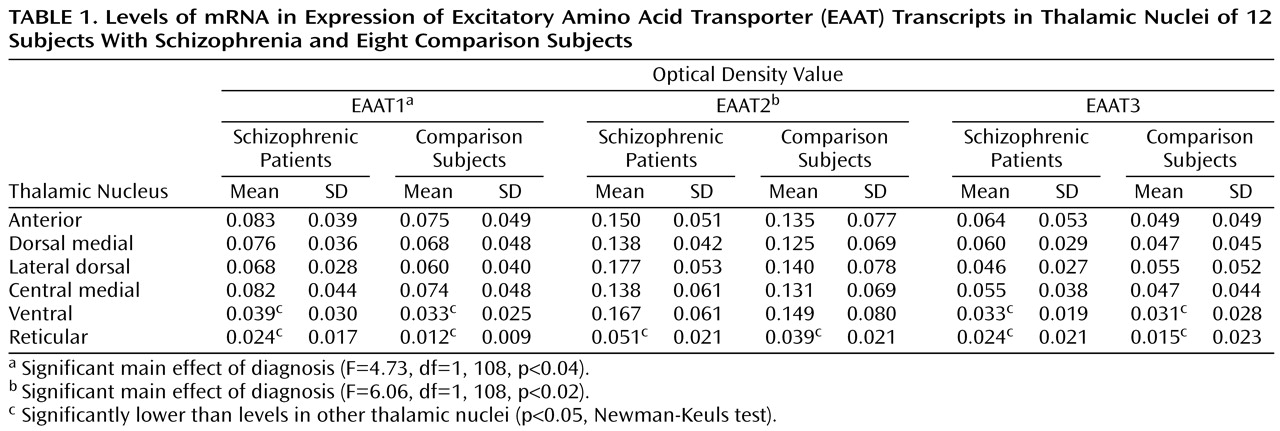
Sense and antisense probes for EAAT1, EAAT2, EAAT3, and EAAT4 transcripts were tested in sections of macaque thalamus; specific labeling was only observed for sections incubated with antisense riboprobe (data not shown). EAAT4 mRNA expression was not distinguishable from background in the macaque thalamus and thus was not included in this study. EAAT1, EAAT2, and EAAT3 were expressed in all nuclei studied (
Figure 1 and
Table 1). As seen in
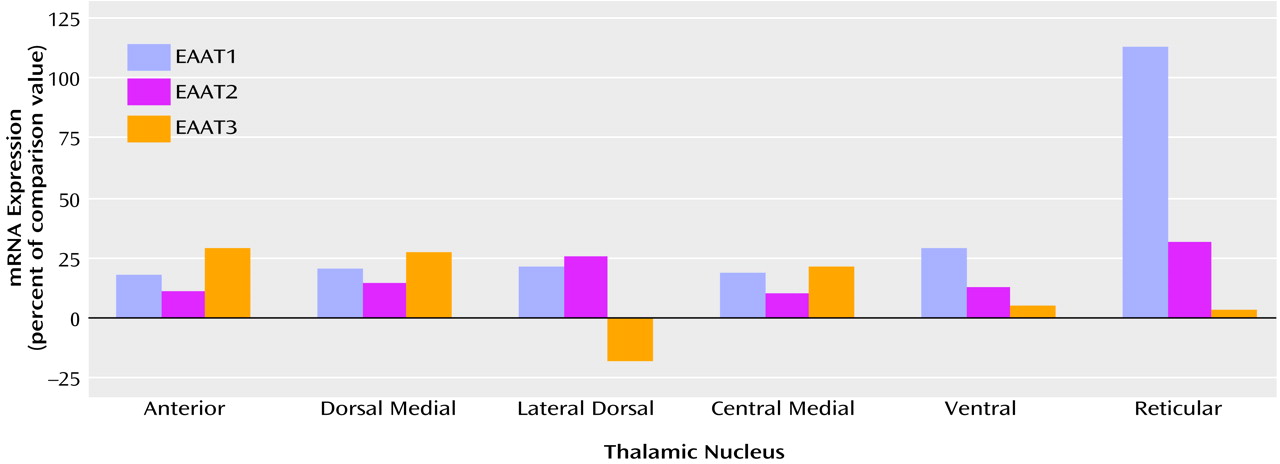
Table 1, there was a main effect of diagnosis for thalamic expression of EAAT1 and EAAT2; there was no main effect of diagnosis for thalamic expression of EAAT3 (F=1.65, df=1, 108, p<0.21). We did not detect any significant diagnosis-by-thalamic nucleus interactions. Post hoc analysis revealed levels of EAAT1, EAAT2, and EAAT3 mRNA expression in the reticular nucleus and EAAT1 and EAAT3 mRNA expression in the ventral nucleus that were significantly lower than the levels of expression in the other thalamic nuclei for both the schizophrenic and comparison subjects. The largest percentage difference in EAAT mRNA expression was observed in the reticular nucleus (
Figure 2). We did not detect a significant relationship between age and excitatory amino acid transporter expression in any thalamic nuclei.
Discussion
In this study, we detected significantly higher levels of EAAT1 and EAAT2 mRNA expression in the thalamus of subjects with schizophrenia, to our knowledge the first such demonstration in this illness. Significant changes in excitatory amino acid transporter expression were not specific to any of the six nuclei examined (no significant diagnosis-by-nucleus interaction), suggesting that small to moderate increases throughout all of the thalamic nuclei examined account for our finding. This result is consistent with the hypothesis that glutamatergic function in the thalamus of schizophrenic subjects is abnormal.
We have previously reported subunit- and binding site-specific deficits in ionotropic glutamate receptor expression in the limbic thalamus in schizophrenia, limited primarily to NMDA receptors
(22,
23). One unifying interpretation of our current and previous data is that an abnormality of presynaptic glutamate release resulting in higher synaptic glutamate levels might stimulate a compensatory increase in glial EAAT1 and EAAT2 expression and decrease expression of postsynaptic NMDA receptors. The juxtaposition of lower NMDA receptor expression and higher excitatory amino acid transporter expression may be further explained by other putative homeostatic regulatory mechanisms
(49). Cytokines, growth factors, protein kinases, phospholipases, and cyclic lipid mediators have all been implicated as factors regulating glutamate reuptake
(50–
55). One mechanism that may directly link NMDA receptor function with excitatory amino acid transporter expression involves arachidonic acid release. In vitro studies indicate that arachidonic acid is released following activation of NMDA receptors and attenuates glutamate uptake in neurons and glia
(56–
63). Such a mechanism might lead to higher levels of excitatory amino acid transporter expression if lower levels of NMDA receptor expression led to less stimulation of arachidonic acid synthesis, thereby disinhibiting excitatory amino acid transporter expression.
Other studies have demonstrated alterations in excitatory amino acid transporter expression in subjects with schizophrenia. A higher ratio of mGluR3/EAAT2 and mGluR5/EAAT2 mRNA expression was detected in the prefrontal cortex of subjects with schizophrenia
(48). In this study, however, there were no significant changes in either EAAT2 or mGluR mRNA expression when not expressed as a ratio. In another study, lower expression of EAAT2, but not EAAT1 or EAAT3, mRNA was detected in the superficial and deep layers of the dorsolateral prefrontal cortex in a cohort of subjects with schizophrenia
(64,
65). Such changes suggest an abnormality in a reciprocal glutamatergic corticothalamic circuit in schizophrenia.
While there are no data evaluating antipsychotic effects on excitatory amino acid transporter expression in the thalamus, the prefrontal cortex and striatum have been studied. In the striatum of rats treated for 30 days with either haloperidol or clozapine, expression of EAAT2 mRNA was lower
(66). Consistent with this finding, in the striatum of rats treated for 27 weeks with haloperidol,
d-[
3H]aspartate uptake was significantly attenuated
(67). Another study demonstrated a decrease in EAAT2 mRNA expression in the cortex of rats following 9 weeks of treatment with clozapine
(68). These data suggest that antipsychotic exposure decreases EAAT expression, suggesting that our finding of elevated expression in schizophrenia is not a drug effect.
Our results demonstrate that alterations in transporter expression in schizophrenia are limited to the glial-based transporters, EAAT1 and EAAT2
(24). Since EAAT3 expression is neuronal and somatodendritic, we speculate that neuronal transporter expression is not affected in thalamic glutamatergic synapses of subjects with schizophrenia
(24,
34,
49). The dramatic effects of GLAST (EAAT1) or GLT-1 (EAAT2) knockout mice, versus the relatively quiescent effects of knocking out EAAC1 (EAAT3), suggest differential functional roles for the transporters that may be reflected in the differential patterns of transporter expression
(31–
39,
42).
Previously, we reported changes in NMDA receptor (NR) subunit and binding site expression in the thalamus of subjects with schizophrenia
(22). The specific findings of alterations in NR
1 and NR
2C subunit expression and binding to the polyamine modulatory site suggested a change in NMDA receptor stoichiometry, while decreases in expression of the requisite NR
1 subunit suggested a deficit in NMDA receptor expression on the cell surface
(22). We now extend our findings of thalamic alterations in schizophrenia to the glutamate transporter family. The finding of higher levels of EAAT1 and EAAT2 expression, taken together with deficits in NMDA receptor expression, suggest differential abnormalities in schizophrenia within families of related molecules. These results highlight the potential importance of examining different components of the glutamate synapse and directly support the notion of thalamic glutamatergic dysfunction in schizophrenia.