Study Design
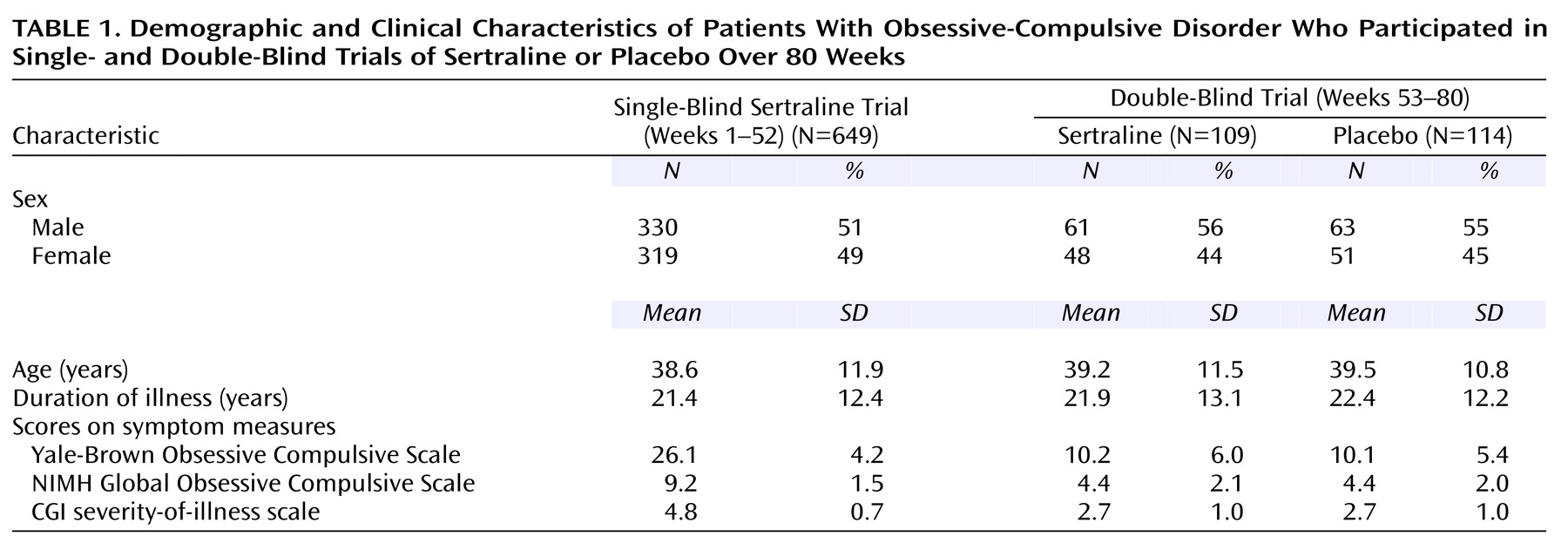
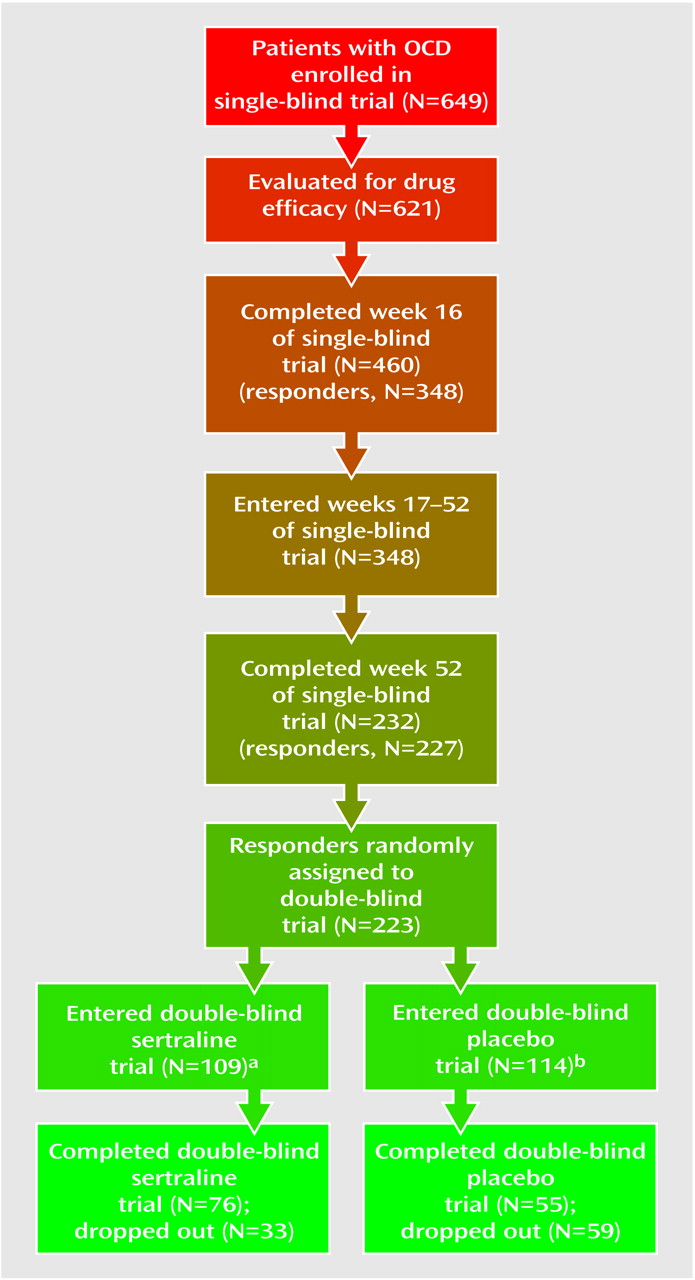
This was an 80-week study conducted at 21 sites in the United States. The subjects were outpatients 18 years of age and older who had a DSM-III-R-defined diagnosis of OCD. At study entry, the patients completed a 1-week drug-free washout period. If they continued to meet study criteria at the end of the washout period (baseline), they entered 16 weeks of single-blind treatment with flexible doses of sertraline. All medication administered throughout the 80-week study period was administered from blister packs holding 50-mg tablets of sertraline (or identical placebo tablets). The patients took 50 mg/day of sertraline during weeks 1 through 4. Patients who failed to respond satisfactorily to treatment could have their dose titrated up to 100 mg/day during weeks 5 and 6 in the absence of dose-limiting adverse events, to 150 mg/day during weeks 7 and 8, and to the maximum dose of 200 mg/day after 8 weeks. In the presence of dose-limiting adverse events, the dose could be reduced in 50-mg/day decrements to a minimum of 50 mg/day. Patients who failed to meet response criteria by the end of 16 weeks of sertraline treatment were withdrawn from the study.
Patients who met the response criteria at the end of week 16 continued with the same dose of single-blind sertraline treatment for an additional 36 weeks; assessments were performed every 4 weeks. For the patients who were not taking the maximum permissible daily dose, an increase in dose was permitted if the investigator thought it might improve the patient’s response. Patients who met relapse criteria on any one of these assessments were scheduled for an assessment 2 weeks later. If they continued to meet the relapse criteria by the 2-week visit, they were scheduled for a third relapse assessment after an additional 2 weeks; if they still met the relapse criteria, they were dropped from the study.
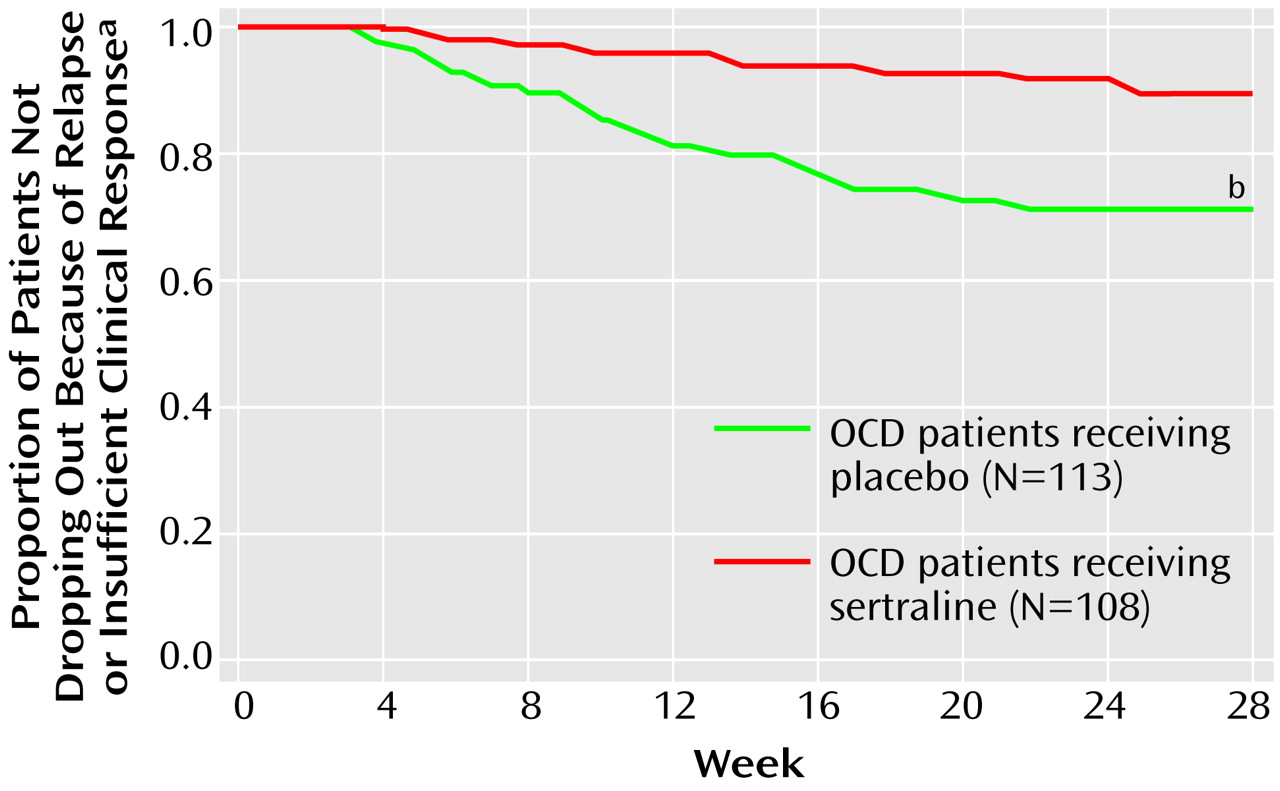
The patients who continued to meet the response criteria at the end of week 52 were randomly assigned to a 28-week double-blind trial with either sertraline or matching placebo. For the patients randomly assigned to sertraline treatment, their daily dose of sertraline as of week 52 was maintained. Until the end of week 56, the patients randomly assigned to placebo took the same number of tablets daily as they had during week 52, but the sertraline dose was blindly decreased by 50 mg/day every 3 days by use of planned substitution of placebo tablets into the blister packs at weeks 53 and 54. Thus, from the end of week 54, these patients took only placebo tablets.
Study Patients
The study patients were recruited by means of newspaper and radio advertisements and from the clinical practices of the investigators. We included male and female outpatients 18 years of age and older who met DSM-III-R criteria for obsessive-compulsive disorder as determined by the Structured Clinical Interview for DSM-III-R (SCID-P)
(18). At baseline, the patients had to have a minimum total score of 20 on the Yale-Brown Obsessive Compulsive Scale
(19,
20) and a score of at least 7 on the NIMH Global Obsessive Compulsive Scale
(21). Patients were excluded from study entry for the following reasons:
1. Having a total score on the 24-item Hamilton Depression Rating Scale
(22) of 17 or higher.
2. Being a woman who was currently pregnant or lactating or of childbearing potential and not using a medically accepted form of contraception.
3. Having a current diagnosis of organic mental disorder, major depression, bipolar disorder, Tourette’s syndrome, or a severe axis II personality disorder or a principle diagnosis of trichotillomania, somatoform disorder, panic disorder, social phobia, or generalized anxiety disorder.
4. Having a current or verified past diagnosis of schizophrenia, delusional disorder, or other psychosis.
5. Having a DSM-III-R-defined diagnosis of alcohol or substance abuse and/or dependence in the past 6 months
6. Having a positive urine drug screening test.
7. Having any medical contraindications to treatment with sertraline.
8. Having a history or evidence of malignancy other than excised basal cell carcinoma.
9. Having an acute or unstable medical illness.
10. Participating in an investigational drug study within 28 days before entering the study.
11. Taking sertraline within 2 months of study entry, not responding to an adequate trial of sertraline in the past, or participating in an investigational study of sertraline.
12. Concomitantly using any psychotropic medication (other than chloral hydrate for sleep).
13. Receiving concurrent behavior therapy for OCD.
14. Receiving treatment with an monoamine oxidase inhibitor within 2 weeks, a depot neuroleptic within 6 months, fluoxetine within 5 weeks, or a neuroleptic, anxiolytic, or antidepressant on a daily basis in the 2 weeks before the first administration of sertraline.
15. Having a test of liver function showing a level at more than twice the upper limit of normal on the first day of the washout period.
16. Being illiterate, or, in the investigator’s judgment, unable or unlikely to follow the study protocol for the full 80 weeks.
The study protocol and forms giving the patients’ informed consent were approved by the institutional review board at each study site. Before study entry, written informed consent was obtained from each subject.
Efficacy Measures
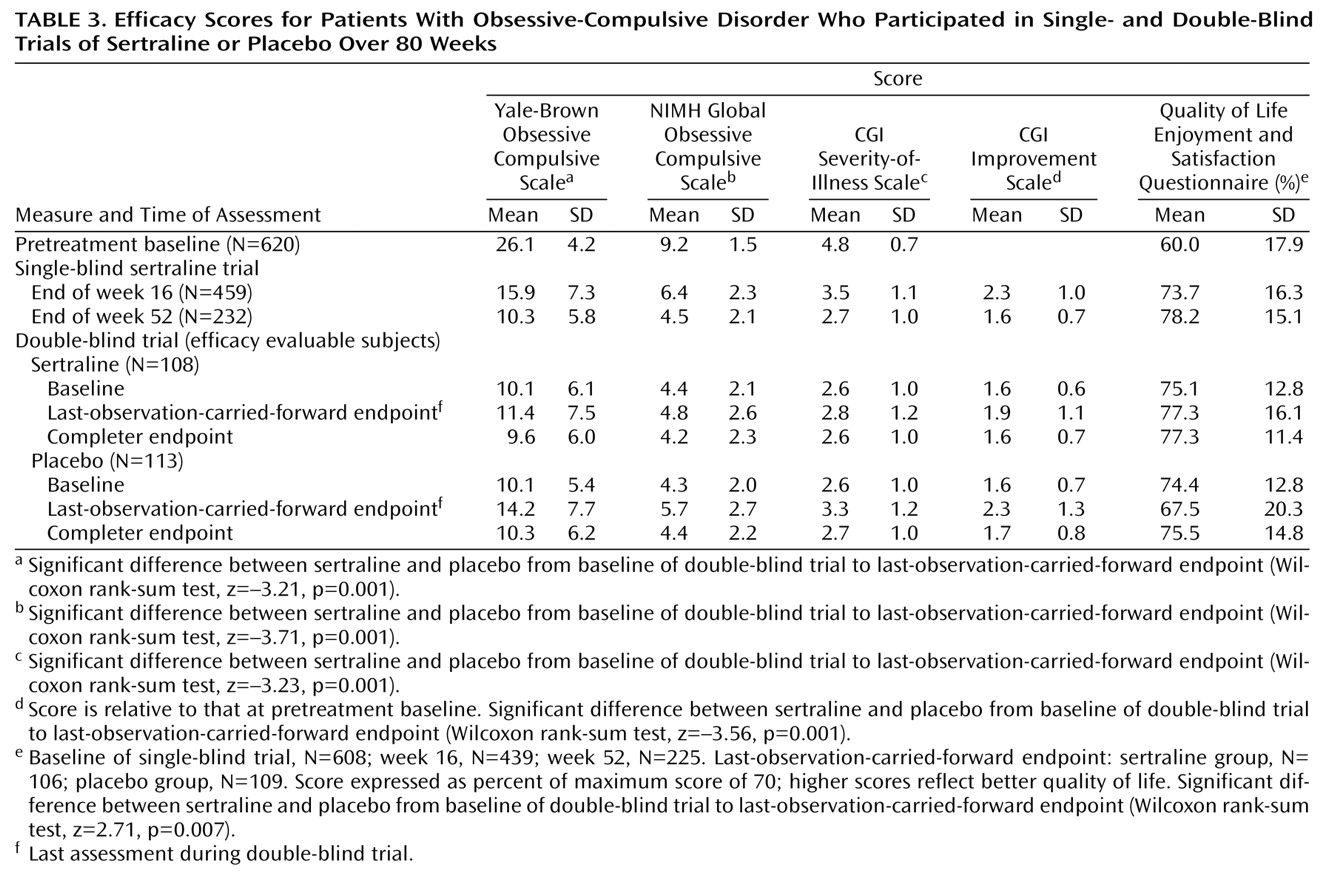
The patients were evaluated at baseline and at subsequent study visits, which occurred weekly for the first 4 weeks and then biweekly until the end of week 16. From week 16 on, the patients were seen and evaluated every 4 weeks unless they met the relapse criteria, in which case an additional assessment was scheduled in 2 weeks. The investigator completed at baseline and at each study visit the Yale-Brown Obsessive Compulsive Scale, the NIMH Global Obsessive Compulsive Scale, the CGI
(23) severity-of-illness scale, and, after the baseline trial, the CGI improvement scale. A patient rating, the Quality of Life Enjoyment and Satisfaction Questionnaire
(24), a scale assessing 16 aspects of quality of life and functioning, was completed at study entry and at study assessments after 16, 52, and 80 weeks of the trial or at study dropout. The investigators specified and agreed to the following definitions during protocol development:
Response
A “response” was defined as occurring when patients’ total scores decreased 25% or more from their baseline (at entry into the single-blind trial) levels on the Yale-Brown Obsessive Compulsive Scale and their CGI improvement scale score was 3 or lower (at least “minimal” improvement). (Although a decrease in score of 25% or more would not be accepted as a “response” criterion in studies involving diabetes, this criterion in commonly used in trials of OCD pharmacotherapy. Its acceptance is testimony to the need for better OCD treatments.) The patients had to meet response criteria by the end of week 16 to continue in the single-blind trial and by the end of week 52 to be randomly assigned to the double-blind phase of the study.
Relapse
After week 16, “relapse” required that a patient meet three criteria during three consecutive visits at 2-week intervals (a period of 1 month):
1. Have an increase in score of 5 points or more (from the week-16 score) on the Yale-Brown Obsessive Compulsive Scale.
2. Have a total score on the Yale-Brown Obsessive Compulsive Scale of 20 or more.
3. And at least a 1-point increase in score on the CGI improvement scale.
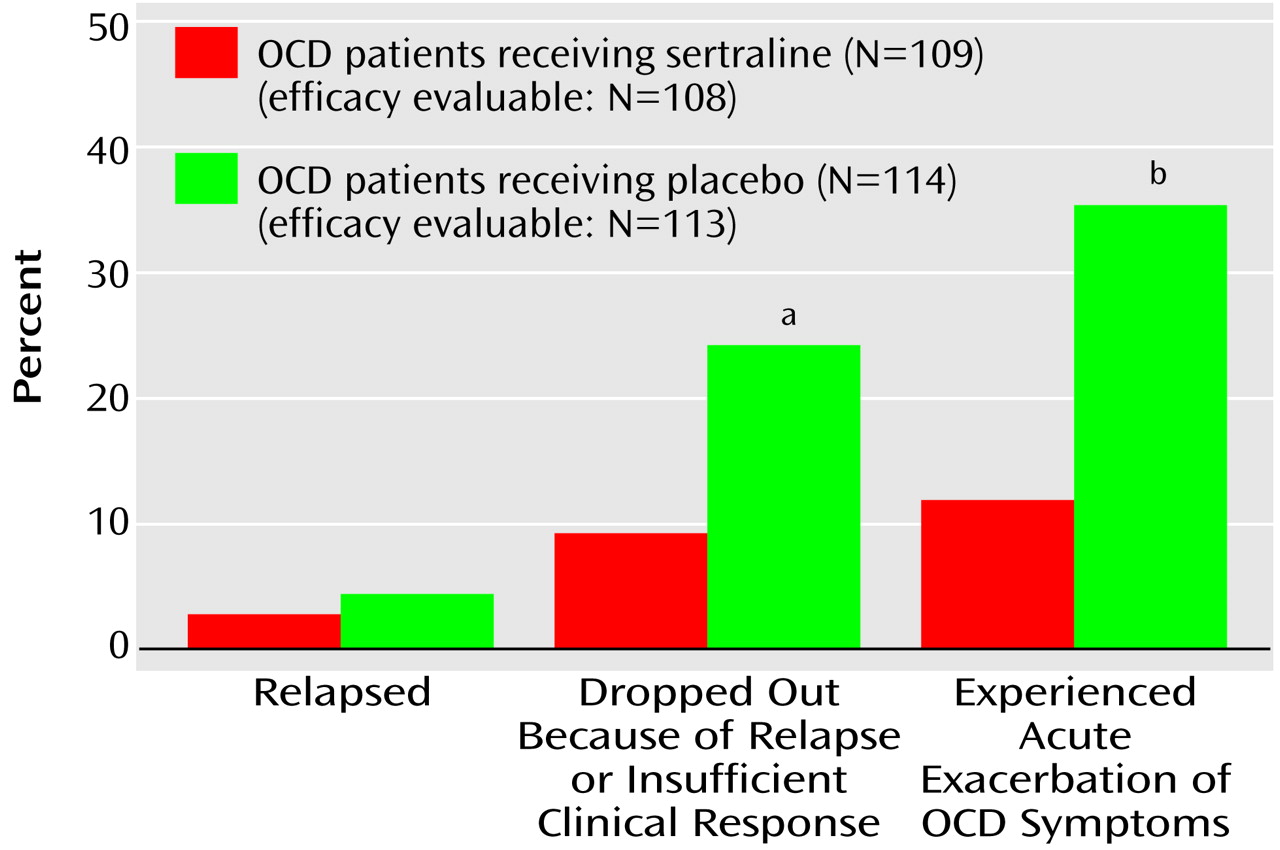
We elected to require 1 month of substantial worsening because OCD patients often experience fluctuations in the intensity of their symptoms. In addition, changes in a patient’s environment may exacerbate their symptoms; e.g., a patient with a fear of germs may have more symptoms if a friend with an acute infection visits his or her home. To differentiate routine oscillations in clinical status from relapse, we used repeated evaluations over a 1-month interval. The patients whose symptoms resolved continued in the study; the patients whose symptoms continued to meet the relapse criteria were classified as relapsed and were automatically removed from the study. The patients could also be removed from the study before completing the 1-month observation period if the investigator believed that it was in the patient’s best interest.
During the double-blind study phase, the definition of relapse was based on the same numeric change in score on the Yale-Brown Obsessive Compulsive Scale and the same change in score on the CGI improvement scale from the end of the single-blind trial (week 52). In many cases the strict protocol definition of relapse was not satisfied because the patient (or investigator) decided, as a result of insufficient clinical response, to drop out of (or drop the patient from) the study immediately at a regular assessment and, consequently, before the three relapse-defining visits.
Acute exacerbation of OCD
In order to describe on a finer scale the outcomes during the double-blind study phase, the investigators wished to have a less stringent, more acute criterion of symptom worsening than the criteria that defined relapse. This endpoint was termed “acute exacerbation of OCD” and used as its reference point the patients’ scores at the end of week 52, which was the baseline for the double-blind phase. Acute exacerbation of OCD was defined as an increase in score of 5 points or more to a minimum of a score of 20 on the Yale-Brown Obsessive Compulsive Scale or an increase in score of at least 2 points on the CGI improvement scale. The patients who met criteria for acute exacerbation of OCD were not automatically released from the study for this reason.
Safety Assessments
A medical history and physical examination were performed on the patients at study entry; a physical examination was repeated at weeks 52 and 80 or when a patient dropped out of the study. Blood pressure, heart rate, and weight were obtained at each study visit.
Both observed and volunteered adverse events were recorded, including date of onset of symptoms, duration, severity, causality, and outcome. ECG testing was performed at study entry and at the end of treatment weeks 2, 10, 16, 32, 52, and 80 (or at study dropout). Laboratory testing was performed at a central laboratory, and blood samples were obtained at each study site at study entry (day 1 of washout) and at the end of treatment weeks 2, 10, 16, 32, 52, 64, and 80 (or at study dropout). Laboratory tests consisted of standard hematology and blood chemistry panels, a urinalysis, and beta human chorionic gonadotropin pregnancy testing for all women of child-bearing potential. Blood samples for T3 uptake and T4 level and urine for a drug screening were obtained only at day 1 of the drug washout.