Psychotic and behavioral disturbances such as hallucinations, delusions, restlessness, disruptive vocalizations, and physical aggression are among the most distressing manifestations of Alzheimer’s disease. They occur with substantial prevalence and have been associated with excess disability, increased caregiver burden, and premature institutionalization
(1,
2). Behavioral disturbances in dementia are heterogeneous, encompassing both agitation occurring in the context of specific delusions or hallucinations and agitation associated with nonspecific factors. Ideal management of these behaviors includes the search for a modifiable environmental or physical precipitant; nevertheless, pharmacotherapy is frequently necessary
(3).
Until recently, conventional antipsychotics were the most frequently prescribed pharmacotherapy for psychosis and behavioral disturbances associated with dementia
(4). Despite the potential advantages of atypical antipsychotics, conventional neuroleptics are still frequently utilized and arguably remain the best-studied medications
(5). A meta-analysis of seven trials that used a double-blind, placebo-controlled, parallel-group design revealed that neuroleptics have a modest overall effect size of 0.18
(6). Previously, perphenazine was extensively used in our hospital to treat behavioral disturbances associated with dementia. Our experience with this conventional neuroleptic suggested that low fixed doses are effective and well tolerated in patients who were not poor metabolizers of the cytochrome P-450 enzyme CYP2D6
(7,
8).
Specific biochemical data have suggested that serotonergic deficits in Alzheimer’s disease contribute to aggressive verbal and physical outbursts, sleep disturbance, depression, and psychosis
(5,
9). Over the past decade, reports on the use of serotonergic medications for the treatment of the behavioral disturbances of dementia have been published
(5). Several case series and one randomized trial
(10) suggested that trazodone is effective. In a more recent study
(11), however, the efficacy of trazodone was not better than that of placebo. In two early placebo-controlled trials, the selective serotonin reuptake inhibitors (SSRIs) alaproclate
(12) and fluvoxamine
(13) were also not found to be more efficacious than placebo. However, the numbers of subjects in these trials were small. In a larger randomized, placebo-controlled trial involving patients with Alzheimer’s disease, 31 patients treated with citalopram showed significant improvement in emotional bluntness, irritability, anxiety, and restlessness
(14). Nonetheless, the subjects in this study were included without regard to possible depressive symptoms. Moreover, behavioral symptoms in this community-based study tended to be mild. The mean pretreatment scores for symptoms of irritability, anxiety, and restlessness were all less than 1.5 on a scale from 0 (absent) to 5 (most severe). In an open pilot study of citalopram, we treated 16 demented patients with delusions or severe agitation who were admitted to geropsychiatric units
(15). Citalopram was well tolerated, and the patients experienced a significant reduction in agitation, hostility, and suspicion. On the basis of these preliminary findings and our earlier experience with perphenazine
(8), we conducted a double-blind, placebo-controlled comparison of citalopram and perphenazine in older patients exhibiting moderate to severe psychosis or behavioral disturbances associated with dementia.
Method
Study Group
All patients with dementia and psychotic or behavioral disturbances who were admitted between October 1995 and January 2000 to the geriatric inpatient units at the Western Psychiatric Institute and Clinic were considered for inclusion in the trial.
To be eligible for the trial, patients had to meet the DSM-IV criteria for a diagnosis of dementia of the Alzheimer’s type, vascular dementia, mixed dementia of the Alzheimer’s type and vascular dementia, or dementia not otherwise specified and not meet the DSM-IV criteria for delirium, schizophrenia, bipolar disorder, or major depressive disorder. The presence of at least one clear “target symptom,” as demonstrated by a score of 3 or more (i.e., moderate to severe) on the Neurobehavioral Rating Scale
(16) agitation items (aggression, agitation, hostility) or psychosis items (delusions, hallucinations, suspiciousness) was also required. Subjects were excluded if there was evidence of clinically significant depression, as demonstrated by a score of 3 or more on the Neurobehavioral Rating Scale depression item. Subjects with unstable physical illness or a neurological illness other than dementia were also excluded. In addition, subjects were required not to have been treated with fluoxetine within the past 4 weeks or with a monoamine oxidase inhibitor within the past 2 weeks. Cognitive enhancers were permitted if the subject had been receiving a stable dose for a minimum of 6 weeks. During the recruitment period, 239 patients were considered for participation. In accordance with the policy of the University of Pittsburgh institutional review board, before approaching potential subjects for consent, the treatment team was consulted to ensure that the research procedures would not be unduly burdensome or disruptive to impending clinical procedures. After the research staff and a physician-investigator had explained the study procedures, goals, and risks to the patient and his or her authorized representative, written informed consent was obtained from the patient’s representative. The patient’s assent was also obtained. Informed consent was obtained for 92 patients, seven of whom were excluded before medications were started. Thus, 85 patients were randomly assigned to treatment groups, took at least one dose of study medication, completed at least one follow-up assessment, and were included in the analysis.
Study Design
The study was a randomized, double-blind, placebo-controlled trial lasting for the duration of inpatient hospitalization, up to 17 days. For better evaluation of the subjects’ behaviors and symptoms, the subjects first completed a 3–5-day observation period and washout of all current psychotropic medications (i.e., antipsychotics, antidepressants, antiepileptics and other mood stabilizers, antiparkinsonian medications, and benzodiazepines) except for lorazepam, which patients could receive during the washout and throughout the study at the lowest possible dose, not to exceed 1 mg/day. Upon completion of the washout, the subjects were randomly assigned to double-blind treatment with 10 mg/day of citalopram for 3 days followed by 20 mg/day for 14 days or 0.05 mg/kg per day of perphenazine for 3 days followed by 0.1 mg/kg per day for 14 days. Medications were administered according to the “double-dummy” technique, twice daily, with citalopram given at 1:00 p.m. and perphenazine at 8:30 p.m. Patients were randomly assigned to receive either citalopram, perphenazine, or placebo according to a 3:3:2 ratio. Early discontinuation from the study was considered in the event of the onset of new symptoms that endangered the safety or health of the patient (e.g., delirium or a serious adverse reaction possibly related to study medication), a significant worsening of psychosis or agitation, an administrative reason (e.g., discharge from the hospital), or when consent was withdrawn. At the conclusion of the study the blind was broken, and the patients were treated by their attending physicians as clinically indicated.
Assessments
On the patients’ admission to the inpatient units, a full diagnostic and laboratory assessment was completed by the clinical team to evaluate for potentially treatable causes of dementia. The patients were also assessed by trained research raters at study enrollment, at the study baseline (i.e., at random assignment), and after receiving study medication for 3, 10, and 17 days (or at discontinuation from the study). Information was obtained from direct observation of the patients, interviews with the patients and nursing staff, and chart review of all notes for the 3 days preceding the rating. Instruments included the Neurobehavioral Rating Scale, Udvalg for Kliniske Undersøgelser (UKU) Side Effect Rating Scale
(17), and the Mini-Mental State Examination (MMSE)
(18). The Neurobehavioral Rating Scale is a 28-item observer-rated instrument derived from the Brief Psychiatric Rating Scale (BPRS) that assesses multiple types of psychopathology. It combines coverage of the breadth of psychopathology addressed in the BPRS with more comprehensive assessment of impairments seen in dementia. Scoring for the Neurobehavioral Rating Scale is based on a 7-point scale of increasing severity (i.e., 0=not present, 1=very mild, 2=mild, 3=moderate, 4=moderately severe, 5=severe, 6=extremely severe). Although the subjects were entered into the study after meeting severity criteria on individual agitation or psychosis “target” symptoms, we have found that a first-order seven-factor model of the Neurobehavioral Rating Scale provides the best fit for correlations among Neurobehavioral Rating Scale items
(19,
20). Consequently, the seven Neurobehavioral Rating Scale factor-based scores (for cognition, agitation/aggression, retardation, depression, apathy, psychosis, and lability/tension) and their sum, yielding a total Neurobehavioral Rating Scale score, were used as our primary efficacy measures. Raters participated in initial training sessions and in retraining sessions yearly. In addition, interrater reliability of the research raters was established and monitored by calculating intraclass correlation coefficients (ICCs) for the Neurobehavioral Rating Scale, UKU Side Effect Rating Scale, and MMSE. Throughout the study, interrater reliability was good to excellent, with ICCs >0.70.
Diagnoses were established according to the DSM-IV criteria, after considering all available information, during a weekly research consensus conference attended by at least three investigator geropsychiatrists and by the research staff. In addition, the criteria for probable or possible Alzheimer’s disease of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association
(21) were used for confirmatory review. Patients were also classified during consensus meetings according to the criteria for the diagnosis of dementia with Lewy bodies
(22). Medical problems were coded on axis III according to the ICD-9-CM classification system.
Statistical Methods
Absolute change in the seven Neurobehavioral Rating Scale factor-based scores was the primary outcome measure. The number of side effects as assessed with the UKU Side Effect Rating Scale, including extrapyramidal symptoms, and use of lorazepam were also compared among treatment groups. Analyses were done on an intent-to-treat basis with the last observation carried forward. Thus, patients who were randomly assigned to a treatment group, received at least one dose of study medication, and completed the baseline and at least one postbaseline evaluation were included in the analyses. All statistical tests were two-tailed. The test for normality (Shapiro-Wilk statistic) indicated that the factor-based scores were not distributed normally. Therefore, differences among the treatment groups in the seven factor-based scores and the percentage change in total, autonomic, and extrapyramidal UKU Side Effect Rating Scale scores were assessed by using the Kruskal-Wallis test. Pairwise comparisons with the Wilcoxon rank-sum test were done when deemed necessary. For total Neurobehavioral Rating Scale scores, which were normally distributed, we performed an analysis of covariance with baseline total scores as covariates. In addition, changes from baseline in the total Neurobehavioral Rating Scale scores were analyzed by using a two-way cross-classification analysis of variance with factors for treatment group and lorazepam use.
Results
Subjects
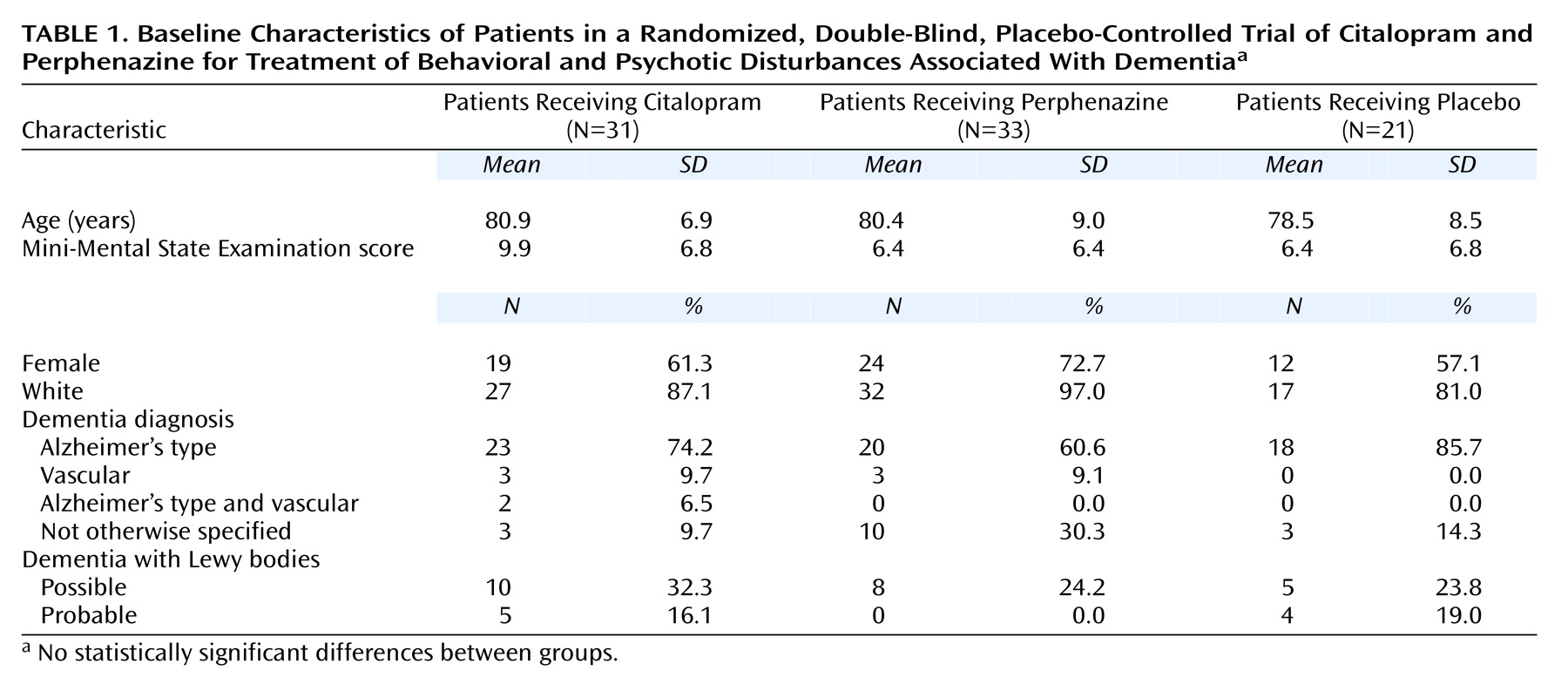
Of the 85 subjects, 61 met the DSM-IV criteria for dementia of the Alzheimer’s type, six for vascular dementia, two for dementia of the Alzheimer’s type and vascular dementia, and 16 for dementia not otherwise specified. Nine of the 85 patients also met the criteria for probable dementia with Lewy bodies and another 23 for possible dementia with Lewy bodies. Thirty-one patients were randomly assigned to receive citalopram (36%), 33 to receive perphenazine (39%), and 21 to receive placebo (25%). One patient who was assigned to receive perphenazine was found on subsequent review to have had a Neurobehavioral Rating Scale depression rating of “moderate.” The results of analyses both including and excluding data for this subject did not differ. Thus all results presented include the data for this subject. The demographic and clinical characteristics of the three treatment groups are presented in
Table 1. The groups did not differ significantly on any measure at baseline.
Patient Disposition
All 31 subjects (100%) assigned to receive citalopram received 20 mg/day; subjects assigned to receive perphenazine received a mean daily dose of 6.5 mg/day (SD=1.7). Thirty-nine patients (46%) completed 17 days of treatment or placebo in the hospital, and 46 (54%) did not. Reasons for discontinuation included possible side effects (N=14), lack of efficacy (N=23), administrative reason (e.g., discharge due to rapid improvement) (N=7), and medication noncompliance (N=2). The three treatment groups did not differ significantly in the overall proportion of patients who discontinued the trial early (citalopram: 16 patients, 52%; perphenazine: 18 patients, 55%; placebo: 12 patients, 57%) (χ2=0.17, df=2, p=0.92) nor in the distribution of reasons for discontinuation (χ2=1.27, df=2, p=0.53).
Efficacy and Safety
Before the start of treatment, the mean total Neurobehavioral Rating Scale scores were 53.5 (SD=10.2), 57.1 (SD=14.0), and 58.3 (SD=11.9) for the citalopram, perphenazine, and placebo groups, respectively. The final mean total Neurobehavioral Rating Scale scores were 43.5 (SD=12.1), 49.9 (SD=14.2), and 56.0 (SD=15.2) for the three respective groups. With the baseline total Neurobehavioral Rating Scale score as a covariate, there was a significant difference in the final total Neurobehavioral Rating Scale scores among the three treatment groups (F=30.8, df=3, 81, p<0.0001). Pairwise comparisons indicated that the final total score of the citalopram group was significantly different from that of the placebo group (t=3.3, df=50, p=0.002), but the final total score of the perphenazine group was not (t=1.5, df=52, p=0.14). On the basis of the total Neurobehavioral Rating Scale scores, the treatment effect sizes for citalopram and perphenazine were 0.64 and 0.36, respectively.
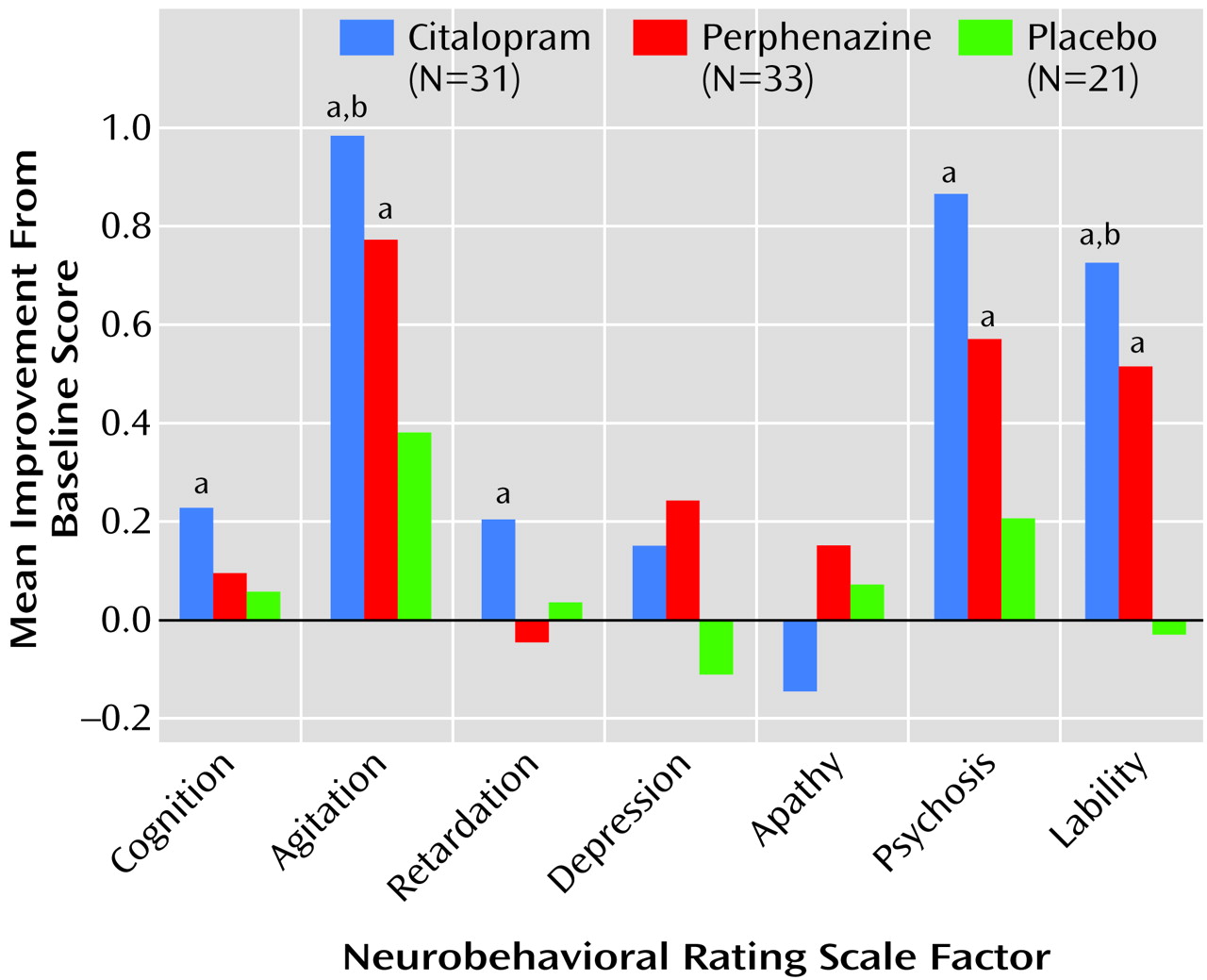
Both the citalopram and perphenazine groups showed significant improvement from baseline with respect to the agitation/aggression, psychosis, and lability/tension factors. The citalopram group also showed significant improvement in the cognition and retardation factors. Those receiving placebo did not demonstrate significant change on any factor (
Figure 1).
The absolute changes in agitation/aggression and lability/tension factor-based scores were significantly different in the three treatment groups (Kruskal-Wallis test χ2=6.7, df=2, p<0.04, and χ2=9.2, df=2, p=0.01, respectively). Pairwise comparisons were significant only for citalopram versus placebo (agitation/aggression factor: χ2=5.0, df=1, p<0.03; lability/tension factor: χ2=9.23, df=1, p=0.002). With regard to side effects, no significant change in total UKU Side Effect Rating Scale score was detected in any of the three treatment groups (F=1.49, df=2, 81, p=0.23). Also, there were no significant differences between groups in autonomic and extrapyramidal symptom subscores (F=0.57, df=2, 61, p=0.57; and F=1.84, df=2, 78, p=0.16, respectively).
Concomitant Medications
Twelve patients who had received a stable dose of donepezil before the current episode of behavioral disturbance continued to receive this medication. Four of these patients were randomly assigned to receive placebo, two to receive perphenazine, and six to receive citalopram. The three treatment groups did not differ significantly with respect to the proportion of patients who required lorazepam for 5 days or more during the trial (citalopram: eight patients, 26%; perphenazine: 11 patients, 33%; placebo: seven patients, 33%) (χ2=0.58, df=2, p=0.83). A two-way cross-classification analysis of variance was performed to determine whether changes in the mean total Neurobehavioral Rating Scale score were associated with the use of lorazepam or treatment assignment and whether there was a interaction between lorazepam use and drug or placebo assignment. A treatment assignment effect (F=4.5, df=2, 79, p<0.02) was seen but no effect of lorazepam use (F=0.53, df=1, 79, p=0.46) and no interaction effect (F=1.9, df=2, 79, p=0.16) were noted.
Discussion
In Alzheimer’s disease patients, psychotic and nonpsychotic behavioral disturbances improved acutely with both citalopram and perphenazine. However, only citalopram demonstrated acute efficacy superior to placebo. At the doses used, both drugs were well tolerated.
The main strengths of this study were the parallel treatment design, the inclusion of a placebo group, and the diagnostic and symptomatic characterization of the subjects, as well as the prospectively defined hypotheses and data analytical plan. It should also be appreciated that the study patients were hospitalized because of the severity of their symptoms, which most likely explains the high attrition rate we observed. Severity of symptoms and the inpatient setting also necessitated a brief trial, the main limitation of the study. The severity of symptoms and brief duration of the trial probably also resulted in a placebo response rate that was considerably lower than that reported in studies with outpatients
(11) or long-term care residents
(23). Nonetheless, for severely affected, hospitalized patients the goal of treatment is to control symptoms rapidly, at least to an extent sufficient to permit discharge, rather than to achieve a full response. Therefore, a trial of 10–17 days fairly represents what can be accomplished during an acute hospitalization, and its results are generalizable to actual practice. Although perphenazine’s effect was modest and not significant relative to placebo, the effect size we observed (0.36) was similar to those reported in other placebo-controlled trials of conventional neuroleptics conducted for longer durations
(6).
Given the brief period of treatment and the severity of symptoms at baseline, the improvement in both overall agitation and specific psychotic symptoms in nondepressed patients with citalopram is notable. There is a need, however, to exercise caution in ascribing an “antipsychotic” effect to an SSRI. Patients were entered into the study if they had symptoms of either psychosis or agitation. Agitation frequently accompanies psychosis, which reduces our capacity to determine the specificity of citalopram for these symptoms. Moreover, psychotic symptoms in Alzheimer’s disease are clearly distinct from those typical of schizophrenia
(24). For example, of the 23 patients experiencing hallucinations in our study, 14 (61%) had only visual hallucinations.
Delusions in demented patients may also result from more generalized impairment, such as misperceptions and impaired judgment in the context of anxiety. Significant relationships have been reported between delusions in Alzheimer’s disease and life events
(25), including marital discord
(26). Therefore, it is possible that “delusions” in Alzheimer’s disease may be attenuated by the anxiolytic effect of an SSRI. Nyth and Gottfries
(14) referred to this action of citalopram as “behavioral stabilization.”
Nonetheless, SSRIs may also have “neuroleptic” effects by reducing dopaminergic outflow
(27), and dysregulation in serotonergic neurotransmission may play an etiologically important role for psychotic symptoms in Alzheimer’s disease patients
(5). For example, common serotonin 5-HT
2A and 5-HT
2C receptor polymorphisms have been associated with visual hallucinations in Alzheimer’s disease subjects
(28), and recently we have found that aggression and psychosis in Alzheimer’s disease subjects were associated with serotonin transporter gene promoter region polymorphisms
(29,
30). Finally, SSRIs differ in their potency and selectivity for serotonin reuptake inhibition, with citalopram being the most selective with moderate potency and high bioavailability
(31). Therefore, future studies should address the relative effectiveness of other SSRIs as well as cross-class comparisons with atypical antipsychotics.