The value of maintenance antipsychotic medication in reducing the risk of psychotic relapse and rehospitalization for patients with schizophrenia has been well established
(1–
3). However, antipsychotic drugs are associated with a variety of adverse effects that can produce subjective discomfort, untoward behavioral effects (e.g., akinesia
[4], akathisia
[5]), and abnormal involuntary movements (e.g., tardive dyskinesia
[6] and tardive dystonia
[7]) and contribute to high rates of noncompliance in medication taking
(8). Although new-generation antipsychotics have lower propensities to cause extrapyramidal side effects, none is yet available in a long-acting injectable formulation, and when long-acting forms do become available, the present data will be important for establishing a benchmark for comparison. In addition, conventional antipsychotics (particularly depot medications) continue to be much more widely used elsewhere in the world than in the United States.
Several long-term studies
(9–
15) have been conducted to determine the impact of substantial reduction of neuroleptic dose in the maintenance phase of treatment. It was hoped that by using the minimum effective dose, cumulative antipsychotic drug exposure could be reduced, with a consequent reduction in adverse effects, particularly tardive dyskinesia. All of these studies used long-acting injectable (depot) antipsychotic drugs. The advantage of depot preparations is that compliance in medication taking is controlled. If a patient relapses, it is clear that the patient is relapsing despite medication, since injections have been given regularly. When oral medication is administered, a relapse might be due to covert noncompliance. In addition, at times it is difficult to reliably determine the sequence of events, i.e., did the patient begin to relapse and then become noncompliant, or did noncompliance precede and, in effect, cause the relapse?
Another advantage of depot medications is that because they are given parenterally, heterogeneity in bioavailability and metabolism is diminished to a considerable extent. Therefore, the correlation between the dose administered and the blood level obtained is significantly higher with depot than with orally administered medication
(16). These factors have important implications in clinical trials in which one of the goals is to determine guidelines for using the minimum effective dose for maintenance treatment. For the routine clinical management of patients with schizophrenia these factors are equally, if not more, important.
Given the widespread use of haloperidol in the short- and long-term treatment of schizophrenia, a controlled trial comparing different doses of haloperidol decanoate was felt to be needed. To our knowledge, this is the only study to date comparing four different doses of any depot (or oral) formulation.
Method
The protocol was approved by the institutional review board of each participating institution. A six-site, double-blind study was conducted to determine the rates of symptomatic exacerbation and adverse effects in patients randomly assigned to four different doses of haloperidol decanoate and treated for 1 year or until relapse. The doses were 25, 50, 100, and 200 mg; each was given intramuscularly once per month.
Study Participants
Inclusion criteria were 1) presence of DSM-III schizophrenia or schizoaffective disorder for at least 2 years, 2) the need for maintenance antipsychotic treatment as evidenced by a history of psychotic decompensation (relapse) when antipsychotic medication was discontinued and reestablishment of relative remission when antipsychotic medication was reinstated, 3) a baseline state of relative remission for at least 3 months during maintenance treatment with antipsychotic medication, and 4) signed statement of informed consent. Patients were excluded if they required treatment with lithium or antidepressants, were women of childbearing potential not practicing acceptable methods of birth control, had a history of or present medical condition contraindicating the use of haloperidol, or had a Brief Psychiatric Rating Scale (BPRS)
(17) rating above a specific threshold on any of four psychotic items, i.e., 4 (moderate) for conceptual disorganization or unusual thought content or 5 (moderately severe) for hallucinatory behavior or suspiciousness. The BPRS items were scored on a scale of 1 to 7.
Randomization was balanced (stratified) for sex and dose of prior maintenance treatment, categorized as <12 or ≥12 mg/day of haloperidol equivalents. Haloperidol decanoate was packaged in ampules containing 0.5, 1.0, or 2.0 ml of either active drug solution (with 50 mg of haloperidol decanoate per milliliter of solution) or placebo. Because the volumes of injectable medication had to vary for the four treatment groups, a nurse not involved in the patient’s management or assessment administered the medication injections. A 2-inch 21-gauge needle and the “z-track” technique were used.
At entry into the study all previous antipsychotic medication was discontinued (patients receiving fluphenazine decanoate were not entered until at least 2 weeks after the last injection; patients receiving haloperidol decanoate entered 1 month after the last injection). During the first 4 weeks of haloperidol decanoate, supplemental oral antipsychotics were allowed, but not afterward. Patients randomly assigned to the 200-mg dose received the 100-mg dose at the first injection, so that assignment to the full four groups did not take place until the second month of treatment. Patients were allowed to receive benztropine mesylate, up to 8 mg/day. Propranolol was permitted for the treatment of akathisia.
Assessment
The study participants were evaluated every 2 weeks for the first 8 weeks, and monthly thereafter, with the BPRS and the Clinical Global Impression scale (CGI)
(18). The SCL-90
(19) was completed at baseline, at weeks 4 and 8, and every 8 weeks thereafter. A modified Simpson-Angus Rating Scale
(20) was completed biweekly for the first 8 weeks and then monthly, and the Abnormal Involuntary Movement Scale (AIMS)
(21) was administered at 12, 24, 36, 48, and 52 weeks. The Social Adjustment Scale
(22) (patient version) was completed at baseline, week 24, and week 48. All raters were trained in the use of the rating scales, and whenever possible the same rater evaluated the patient throughout the trial. Formal reliability studies were not required.
Symptomatic exacerbation was defined by increased rating scale scores calibrated to a predetermined threshold for each subject on the basis of baseline ratings on the four psychotic items of the BPRS (unusual thought content, conceptual disorganization, hallucinations, suspiciousness). An increase on one psychotic item of at least 2 scale points was required for a patient to be considered as worse, with the exception of patients who at baseline scored “not present” on conceptual disorganization or hallucinatory behavior, in which case an increase of 3 scale points was required, or patients who scored “not present” or “mild” on suspiciousness, in which case an increase of 4 or 3 scale points, respectively, was required. Whenever worsening occurred, patients were withdrawn from the study and treated as clinically appropriate. Blood samples were obtained from a subset of patients, and plasma levels were determined (data not presented).
Results
Of the 119 patients who entered the study, 105 completed the double-blind, 1-month titration phase. Patients who did not complete this phase were excluded from further analyses; four were in the 25-mg group, two were in the 50-mg group, five were in the 100-mg group, and three were in the 200-mg group.
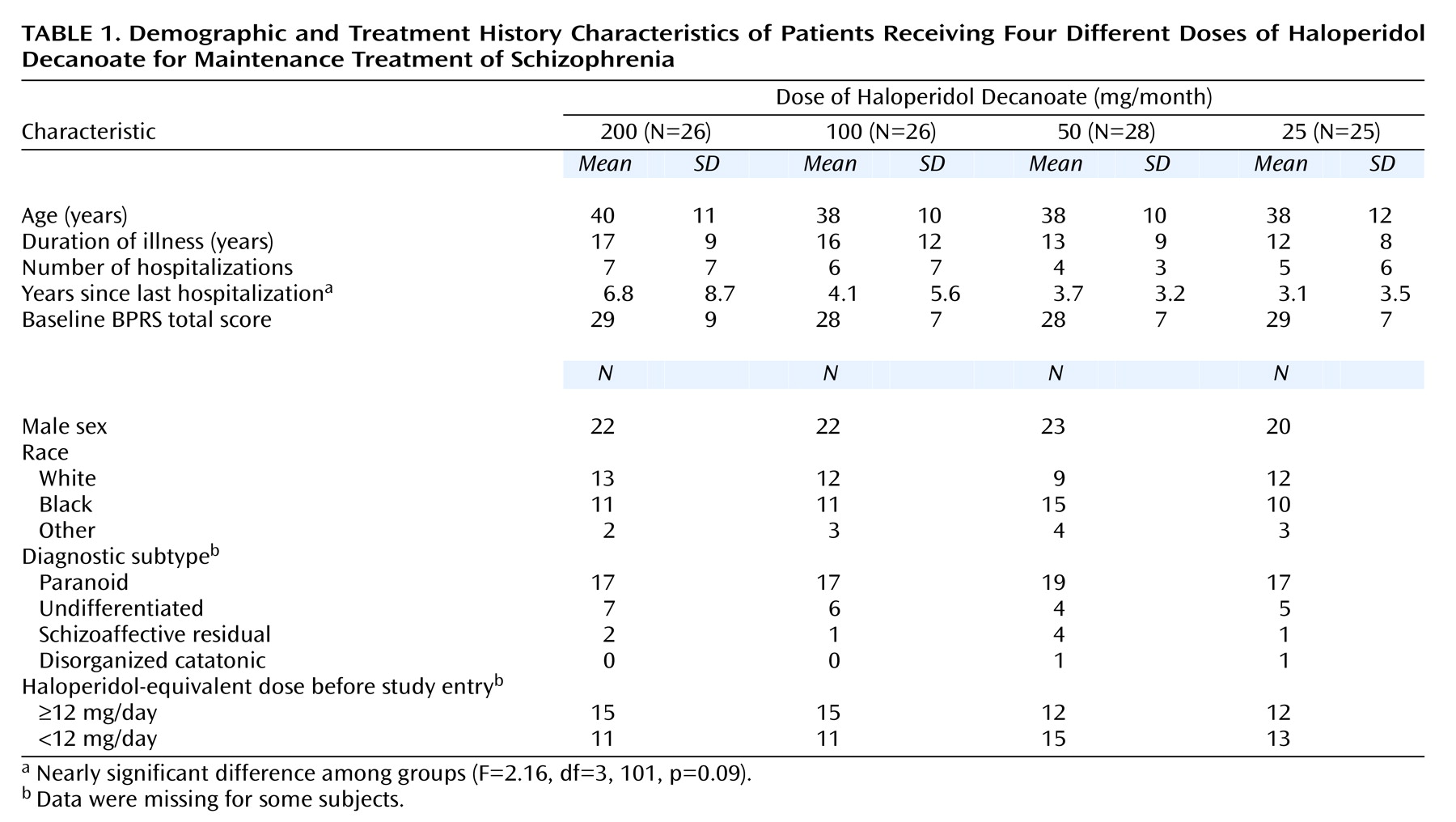
Demographic and treatment history characteristics are provided in
Table 1. Overall, the sample was predominantly male, with equal representations of Caucasians and African Americans. The mean age at study entry was 39 years, and the patients had been ill for 15 years on average and had a mean of six previous hospitalizations. The interval between last hospitalization and entry into the study was, on average, over 4 years. Most of the subjects were diagnosed as having the paranoid subtype. All of the patients were outpatients.
Exacerbations
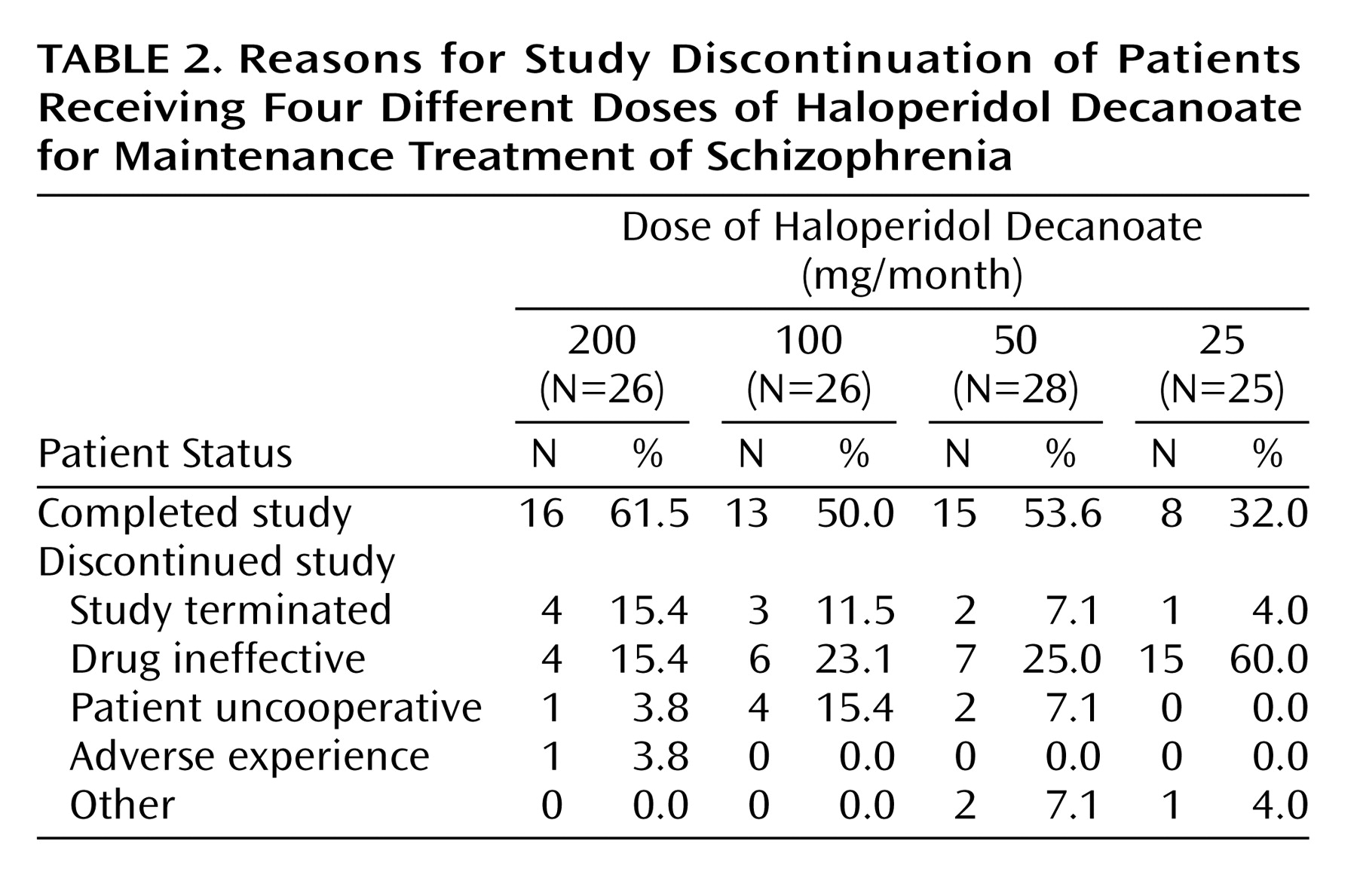
Reasons for discontinuation from the trial are presented by treatment group in
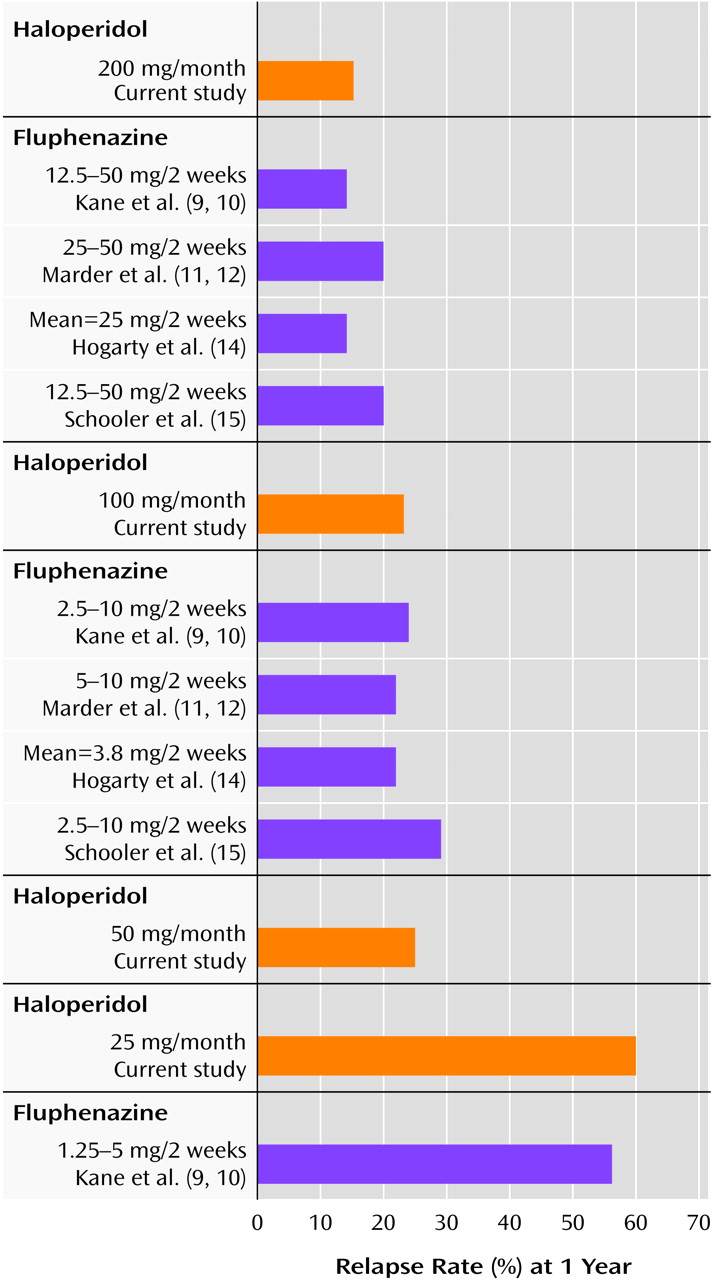
Table 2. Of the 26 patients assigned to the 200-mg group, four (15.4%) experienced a symptomatic exacerbation. In contrast, 15 (60.0%) of the 25 patients receiving 25 mg experienced an increase in symptoms. For the 100-mg and 50-mg groups, the rates were 23.1% and 25.0%, respectively. (The study was terminated by the sponsor for administrative reasons before the blind was broken and before all subjects had completed the trial; 10 patients did not complete the trial for that reason.)
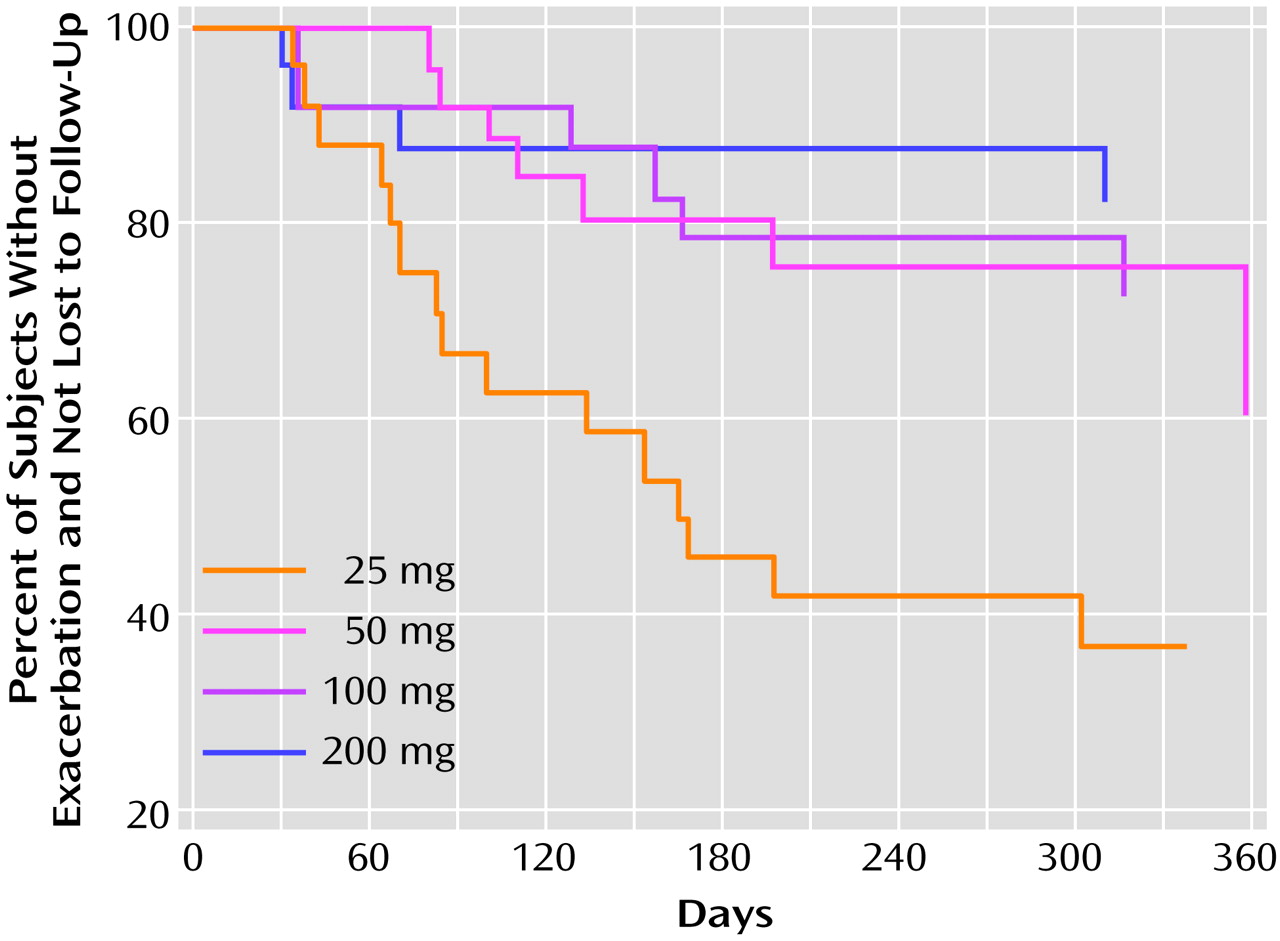
In order to test for differences between doses in time to exacerbation, survival analyses were conducted by means of the proportional hazards survival regression, or Cox model
(23). The analyses included patients who met the criteria for exacerbation plus the patients who were lost to follow-up without documented worsening, who were considered nonrelapsed through their last visit. Survival curves are presented in
Figure 1.
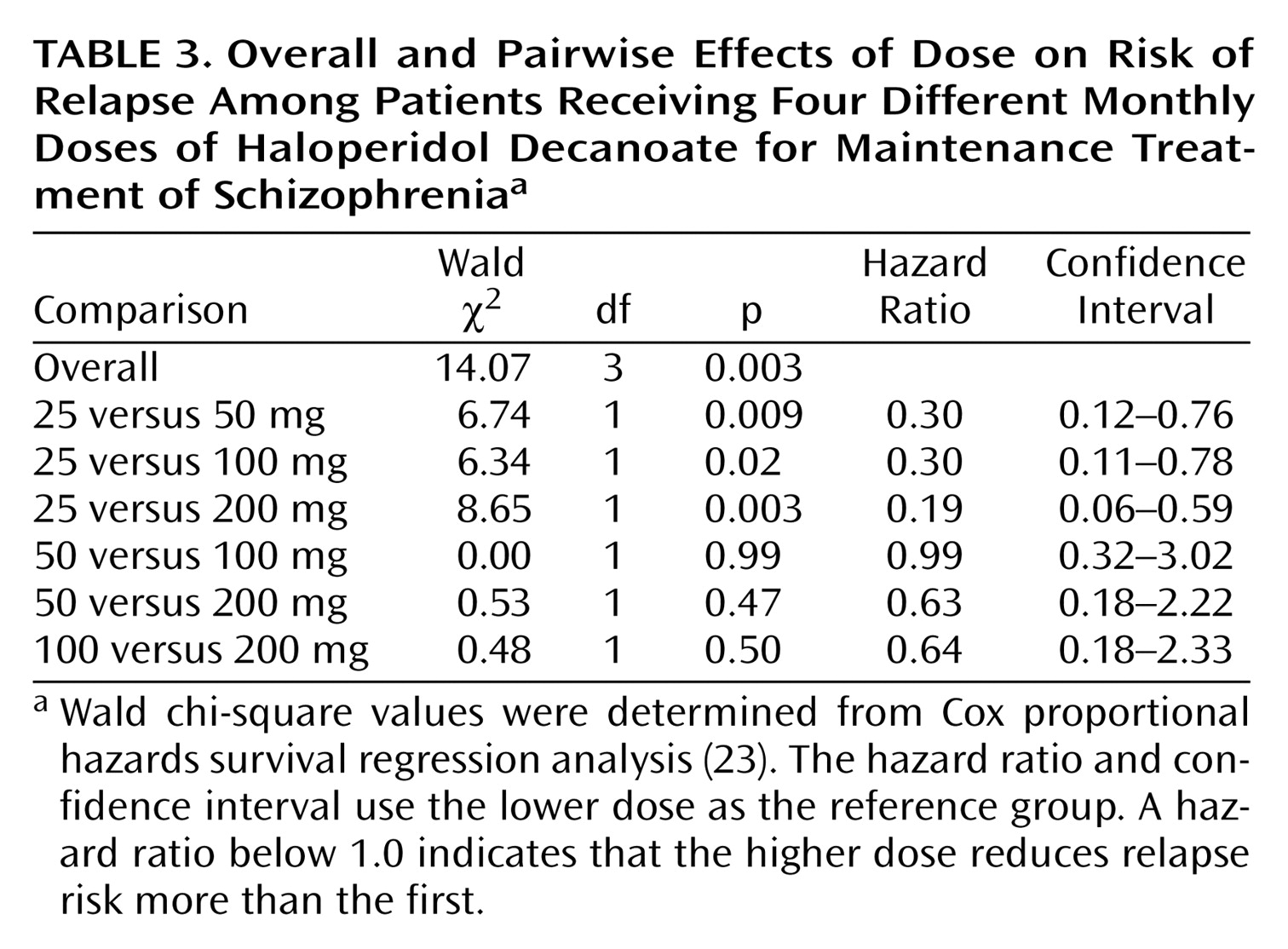
Wald chi-square and p values for the analysis of the data on exacerbation are presented in
Table 3 for the overall and pairwise comparisons. Hazard ratios and confidence intervals are also provided. The association between increasing dose and increasing probability of remaining in the study without relapse was statistically significant. Pairwise comparisons showed the probability to be significantly greater for the 200-mg, 100-mg, and 50-mg groups than for the 25-mg group. No other pairwise comparisons were significant.
Covariate analyses of time to worsening were carried out by using years since last hospitalization, duration of illness (in years), and number of previous hospitalizations. These covariates were selected because their values tended to be greater for the high-dose group at baseline; however, only years since last hospitalization approached statistical significance (p<0.09). When years since last hospitalization was used as a covariate, the overall effect of increasing dose on time to relapse was marginally significant (p=0.06). Using duration of illness as a covariate gave the same results as the analysis of variance with the exception that the pairwise comparison of the 25- and 100-mg groups was only marginally significant. The association between increasing haloperidol dose and increasing probability of not relapsing remained statistically significant when number of previous hospitalizations was used as a covariate.
In comparing the four treatment groups on measures of psychopathology, we found no significant differences at baseline on total BPRS score. There were significant effects of dose on the analyses of covariance (ANCOVAs) at endpoint (i.e., final ratings available for all patients, including those experiencing exacerbations) for the BPRS thought disturbance factor score, the total BPRS score, and the CGI severity and change scores. The patients receiving the lowest dose scored higher than those receiving the larger doses (results of other analyses, including the anergia and anxiety/depression factors, were not significant). There were no significant pairwise differences between the higher-dose groups. There was no significant difference on any measure at week 52 for patients who remained in the study for the entire year.
Scores on the patient version of the Social Adjustment Scale
(22) at baseline and at 24 and 48 weeks were available for the majority of patients. There was no baseline difference on this measure and no difference at 24 weeks, 48 weeks, or endpoint across the dose groups, according to ANCOVA.
No differences between treatment groups were found on any of the six SCL-90 factors at baseline, at week 24, or at week 48 or endpoint.
Higher baseline scores on the BPRS thought disorder factor were correlated with relapse (Wald χ
2=6.77, df=1, p=0.009). In a proportional hazard survival regression analysis
(23) the difference between 25 mg and all other doses remained significant even when this baseline variable was included as a covariate. No other baseline measures were correlated with relapse, including prestudy oral medication dose and gender. As previously indicated, the majority of patients were male, as is the case with many such studies. We were therefore unlikely to identify any potential sex differences in response to specific doses; however, in general there is no evidence of a sex effect on dose-response relationships in maintenance treatment. The significance of the results was equivalent when prestudy dose, gender, and site were included as main effects covariates in the models.
Adverse Effects
There was no significant difference between treatments in the number of patients who left the trial because of adverse effects (only one patient taking 200 mg was in this category). The total scores on the AIMS and Simpson-Angus Rating Scale were square-root transformed to normalize the distribution. ANCOVAs were conducted with baseline values as covariates. There was a significant difference in total score on the Simpson-Angus scale at 1 year (F=3.21, df=3, 45, p=0.03) but not at endpoint (F=0.06, df=3, 95, p=0.98); at 1 year the score for the 25-mg group was significantly lower than the score for the 200-mg group, but there were no significant differences among the three higher-dose groups.
On the AIMS there was a significant difference across groups in severity of abnormal involuntary movements at the 1-year assessment (ANCOVA: F=3.22, df=3, 50, p=0.03) but not at endpoint (F=1.54, df=3, 99, p=0.21). The only pairwise difference was between 100 and 200 mg both at 1 year (t=3.02, df=50, p=0.004) and at endpoint (t=2.13, df=99, p=0.04). The fact that differences between the 200-mg group and either the 50-mg or 25-mg group were not significant suggests that we cannot conclude greater risk is associated with a higher dose, although for evaluating the risk of tardive dyskinesia, this is a small group followed for a short time.
In addition to comparing scores on the Simpson-Angus Rating Scale, we also contrasted the use of anticholinergic medications and β blockers. There were no significant differences in the use of antiparkinsonian medication, measured as either the proportion of time in the study that a subject received antiparkinsonian medication (F=1.61, df=3, 75, p=0.19) or the mean daily dose of anticholinergic medication administered (F=0.27, df=3, 75, p=0.84). Nor did we find any difference between groups in the use of β blockers to treat akathisia.
Discussion
The results of this study should be viewed in the context of previous results with dose comparisons for fluphenazine decanoate (
Figure 2). Kane et al.
(9,
10) reported results from a 1-year random-assignment study of three different fixed doses of fluphenazine decanoate: 12.5–50 mg, 2.5–10 mg, and 1.25–5 mg, each given every 2 weeks. The cumulative relapse rate among patients receiving the very low dose was 56%, compared to 24% for the low dose and 14% for the standard dose. Despite a significantly higher relapse rate among the patients receiving the very low dose, most of the subjects who did relapse were readily restabilized after a temporary dose increase and did not require rehospitalization. Patients receiving the very low dose did better on some measures of psychosocial adjustment than patients receiving the standard dose
(24). An analysis of early signs of tardive dyskinesia showed a significant advantage for the very low dose at the end of the 1-year trial
(10).
Marder et al.
(11,
12) randomly assigned 66 male outpatients to 5–10 mg or 25–50 mg fluphenazine decanoate given every 2 weeks. The patients were followed for up to 2 years and continued to take the assigned fixed dose of 5 or 25 mg as long as they maintained their baseline level of remission. The two treatments produced similar, nonsignificantly different relapse rates after 1 year, 22% with 5–10 mg and 20% with 25–50 mg, and after 2 years the rates were 44% with the lower dose and 31% with the higher dose.
Hogarty et al.
(14) randomly assigned 70 stable outpatients to receive a standard dose of fluphenazine decanoate (mean=25 mg every 2 weeks) or a minimal dose representing approximately 20% of the dose usually prescribed (mean=3.8 mg every 2 weeks). At the end of 1 year the relapse rate for the patients receiving the standard dose was 14%, and the rate for the low-dose group was 22%. After 2 years the relapse rates were 24% and 30%, respectively.
Schooler et al.
(15) studied 313 stable outpatients who were randomly assigned to a standard dose of fluphenazine decanoate (12.5–50 mg every 2 weeks), a low dose (2.5–10 mg every 2 weeks), or a targeted intermittent strategy (vehicle only, every 2 weeks). At the end of 1 year the relapse rates, based on criteria similar to those used in the present study, were 20% for the standard dose and 29% for the low dose. This trial, like the studies of Marder et al.
(11,
12) and Hogarty et al.
(14), lasted for 2 years. By 2 years the rates had risen to 23% and 36%, respectively. Thus, there appears to be an increased risk of relapse with a reduced dose over time.
The results of the present study suggest that the 200-mg/month dose of haloperidol decanoate is associated with the fewest relapses. Patients assigned to this group had a relapse rate (15%) that compares favorably to the best results in maintenance trials with standard doses of fluphenazine decanoate (
Figure 2). In addition, there was no substantial evidence that the good results for relapse prevention with 200 mg were associated with a greater risk of adverse effects or subjective discomfort. According to objective ratings of extrapyramidal symptoms, the 25-mg group had lower scores, but there were no significant differences in mean duration or mean daily dose of anticholinergic treatment. At the same time, the rates of symptom exacerbation for 100 mg (23%) and 50 mg (25%) were not significantly greater than that seen with 200 mg. The consequences of symptom recurrence in this study were generally not severe, and no significant differences were observed in terms of psychosocial adjustment between patients receiving 25 mg (60% experienced exacerbations) and the other treatment groups. If there was clear evidence that the level of adverse effects was significantly lower in the 100-mg or 50-mg group, the lower dose would have an advantage in terms of the risk-benefit ratio. If a dose reduction is not associated with a significantly lower risk of adverse effects and/or noncompliance, there is little justification for the lower dose if any increase in the rate of relapse or exacerbation is likely. On the basis of these data, dose reduction is difficult to justify, but the longer-term implications cannot be addressed with the current design. Without long-term data and a larger group of subjects it is difficult to draw conclusions regarding relative risk for tardive dyskinesia, but in general few studies have been able to demonstrate a dose-response relationship for tardive dyskinesia. It is also possible that higher doses are more likely to suppress underlying abnormal involuntary movements that would be detectable only after dose reduction or discontinuation.
These results are important in terms of adding needed dose-response data on haloperidol decanoate to those available on fluphenazine decanoate. To our knowledge, this is the only comparison of four different doses of a single long-acting injectable drug. The doses were chosen to represent discrete alternatives within a clinically relevant range. Since haloperidol decanoate and fluphenazine decanoate have different pharmacokinetic properties, these data are particularly helpful in establishing what appear to be comparable maintenance doses in terms of overall efficacy (
Figure 2).
It should be emphasized, however, that these studies were conducted by using somewhat different methods. At the same time it is striking how comparable the results are for patients receiving conventional doses.
The lack of increased adverse effects at the highest dose of haloperidol (200 mg) is notable. It has been suggested
(25,
26) that the risk of developing extrapyramidal side effects with fluphenazine decanoate is one and a half times as high as that associated with oral neuroleptics, whereas with haloperidol decanoate the risk appears to be 25% less than in comparison groups taking oral forms. These analyses are crude and may be influenced by a variety of different factors, however, including problems in establishing true dose equivalence. The fact that haloperidol pharmacokinetics appear better suited to a once-monthly injection interval than is fluphenazine decanoate may also be relevant. However, Carpenter et al.
(27) found no significant differences in relapse, symptoms, or side effect measures in 55 patients randomly assigned to fluphenazine decanoate (25 mg) given every 2 weeks or every 6 weeks over 1 year. These data suggest that with fluphenazine decanoate the injection interval can be lengthened without significant increase in relapse rate, at least during the first year.
These results provide some guidelines as to the effective doses of haloperidol decanoate. These data, however, should be used only as a guide, and the ultimate decision about the optimum dose for a given patient should be based on a variety of individual and environmental factors, including but not limited to the nature and severity of previous relapses, environmental stressors and support, vulnerability to adverse effects, and awareness of and response to prodromal signs of relapse.
Since a relapse resulting from too low a dose may not occur for weeks or months, titration in maintenance treatment can be difficult. Sixty percent of the patients in the 25-mg/month group in this study experienced a symptom exacerbation within 1 year, but for half of these patients, the exacerbation occurred more than 50 days after the 25-mg dose was instituted. It is hoped that data such as those presented here can provide valuable guidance. It is important to emphasize, however, that translating findings involving the dose-response relationship for depot medications to dosage guidelines for oral medication is difficult and that without studies specifically designed to establish relative clinical equivalence, such extrapolations should be made with caution.
The choice of the doses used in this study was based on starting with a standard reference dose (i.e., 200 mg) and then examining three clearly discreet lower doses in order to determine by means of a controlled comparison the degree to which dose reduction is desirable.
It is possible that an even higher dose could have been associated with a lower exacerbation rate, but the outcome in the 200-mg group was as good as in any other 1-year study of depot drugs of which we are aware.
Acknowledgments
At Hillside Hospital the participants were Margaret Woerner, Ph.D., Stavros Sarantakos, M.D., Blair Skolnick, M.D., Dominick Gadaleta, M.D., Sabina Meyer, and Abby Freinberg, R.N. At the University of Pittsburgh/Mayview State Hospital, the participants were Robert Conley, M.D. (initial principal investigator), Robert W. Baker, M.D., Jane Collins, B.S.N., and Joyce Bell Delaney, B.A., R.N. At the University of Miami School of Medicine/VA Medical Center the participants were Ramon Boza, M.D., and staff of the Mental Hygiene Clinic, and at the Jackson Memorial Hospital/Psychiatric Outpatient Clinics, Carol Sirvent, R.N., participated. At the Psychiatric Institute/University of Illinois at Chicago the participants were Mohammed Hassan, M.D., and Benjamin Grerl, M.D. At the West Los Angeles VA Medical Center, Joanne McKenzie, R.N., participated, and at the Los Angeles County/University of Southern California Medical Center the participants were Lawrence Gross, M.D., Claude Arbour, M.D., Karl Burgoyne, M.D., and Ralph Koek, M.D. At Oregon Health Sciences University the participants were Thomas Hansen, M.D., William Hoffman, M.D., and Jenelle Fleck, R.N., M.N.