The dopamine hypothesis of bipolar disorder implicates dopamine hyperactivity in mania. The most robust evidence for this hypothesis is derived mainly from observations of the behavioral effects of dopamine agonists and antagonists. For example, amphetamine, a psychostimulant, produces euphoria and other behavioral effects that are very similar to mania
(1–
3); these responses can be blocked by dopamine antagonists
(4–
7). There is evidence that dextroamphetamine-induced arousal and euphoria in humans is mediated by a dopaminergic mechanism, as it is blocked by the selective dopamine blocker pimozide
(8). Administration of drugs that increase dopamine transmission, such as
l-dopa,
d-amphetamine, piribedil, and bromocriptine, has been reported to precipitate mania in patients with bipolar depression
(9–
14).
Alpha-methyl-para-tyrosine, which blocks tyrosine hydroxylase and decreases dopamine synthesis, is effective in reducing manic symptoms
(15), whereas fusaric acid, which inhibits dopamine β-hydroxylase and raises dopamine levels, worsens manic symptoms
(16). Conventional neuroleptics such as chlorpromazine, haloperidol, and trifluoperazine are all very effective in treating mania
(17), and all conventional neuroleptics share the property of blocking dopamine receptors and thereby decreasing dopamine transmission. That the efficacy of neuroleptics is related to dopamine receptor blockade is suggested by the similar efficacy of pimozide, a more specific dopamine receptor antagonist, in reducing manic symptoms
(18,
19). Further support for the dopamine hyperactivity theory of mania is provided by the observation that the
cis isomer of clopenthixol, which possesses D
2 receptor blocking properties, is effective in treating mania while the
trans isomer, which does not block D
2 receptors, has no antimanic properties
(20).
Studies that have attempted to provide direct evidence for dopamine hyperactivity in mania have yielded inconsistent results. For example, the levels of homovanillic acid (HVA), a dopamine metabolite, have been reported to be higher
(21–
28) or no different
(29,
30) in the CSF of manic patients than in the CSF of comparison subjects. Similarly, probenecid-induced accumulations of HVA have been reported to be no different
(27–
29,
31) or higher
(32) in manic subjects than in comparison subjects. Studies of growth hormone responses to apomorphine challenge have also yielded inconsistent results, with some researchers reporting no difference
(33) and others reporting higher
(34) or lower levels
(35) in manic patients, relative to comparison subjects.
Positron emission tomography (PET) can be used to examine various aspects of dopamine function in humans. This technique has been used widely to assess dopamine function in acute schizophrenia
(36,
37). To our knowledge, just one study has used PET to measure dopamine function in acute mania
(38), although two recent studies used single photon emission computed tomography (SPECT)
(39) and PET
(40) to assess aspects of presynaptic dopaminergic function in euthymic bipolar disorder patients. Pearlson and colleagues
(38) examined dopamine D
2 receptors using PET and [
11C]
N-methylspiperone in patients with bipolar disorder, compared with normal subjects and neuroleptic-naive schizophrenic patients. D
2 receptor density was higher in seven patients with bipolar disorder who were psychotic and in 10 schizophrenic patients than in seven nonpsychotic patients with bipolar disorder and the normal comparison subjects. The authors concluded that the higher number of D
2 receptors is a marker of psychosis and is not specific to schizophrenia or other disorders. However, it is noteworthy that this study included only five nonpsychotic manic patients (the other two were depressed), and, of these five, two had Young Mania Rating Scale scores of 9 and 5, suggesting minimal symptoms. Therefore, the question of whether or not higher dopamine D
2 receptor density is associated with acute mania is unresolved.
In this study, we assessed presynaptic dopamine function in neuroleptic- and mood-stabilizer-naive nonpsychotic manic patients before and after treatment with divalproex sodium by measuring [18F]6-fluoro-l-dopa ([18F]DOPA) uptake in the striatum.
Results
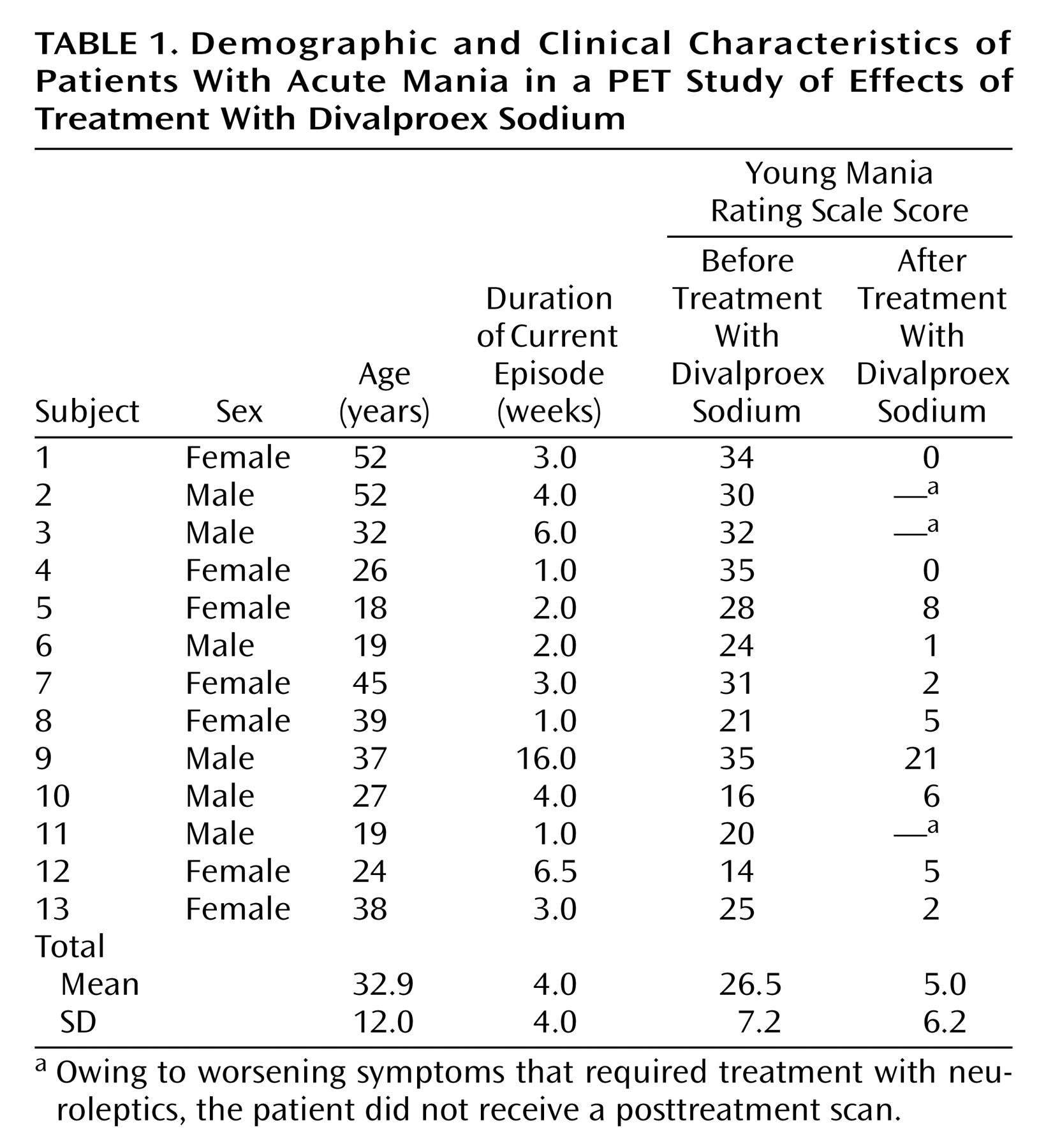
A total of 13 patients (seven women and six men) and 13 healthy comparison subjects (seven women and six men) participated in the study. Demographic and clinical characteristics of the patients are presented in
Table 1. The mean age of the patients was 32.9 years (SD=12.0, range=18–52), and the mean age of the comparison subjects was 31.6 years (SD=11.5, range=20–51). There was no significant difference in age between the patient group and the comparison group (t=0.28, df=24, p=0.77). The patients’ mean Young Mania Rating Scale score was 26.5 (SD=7.2) before treatment and decreased to 5.0 (SD=6.23) after 2–6 weeks of divalproex sodium treatment.
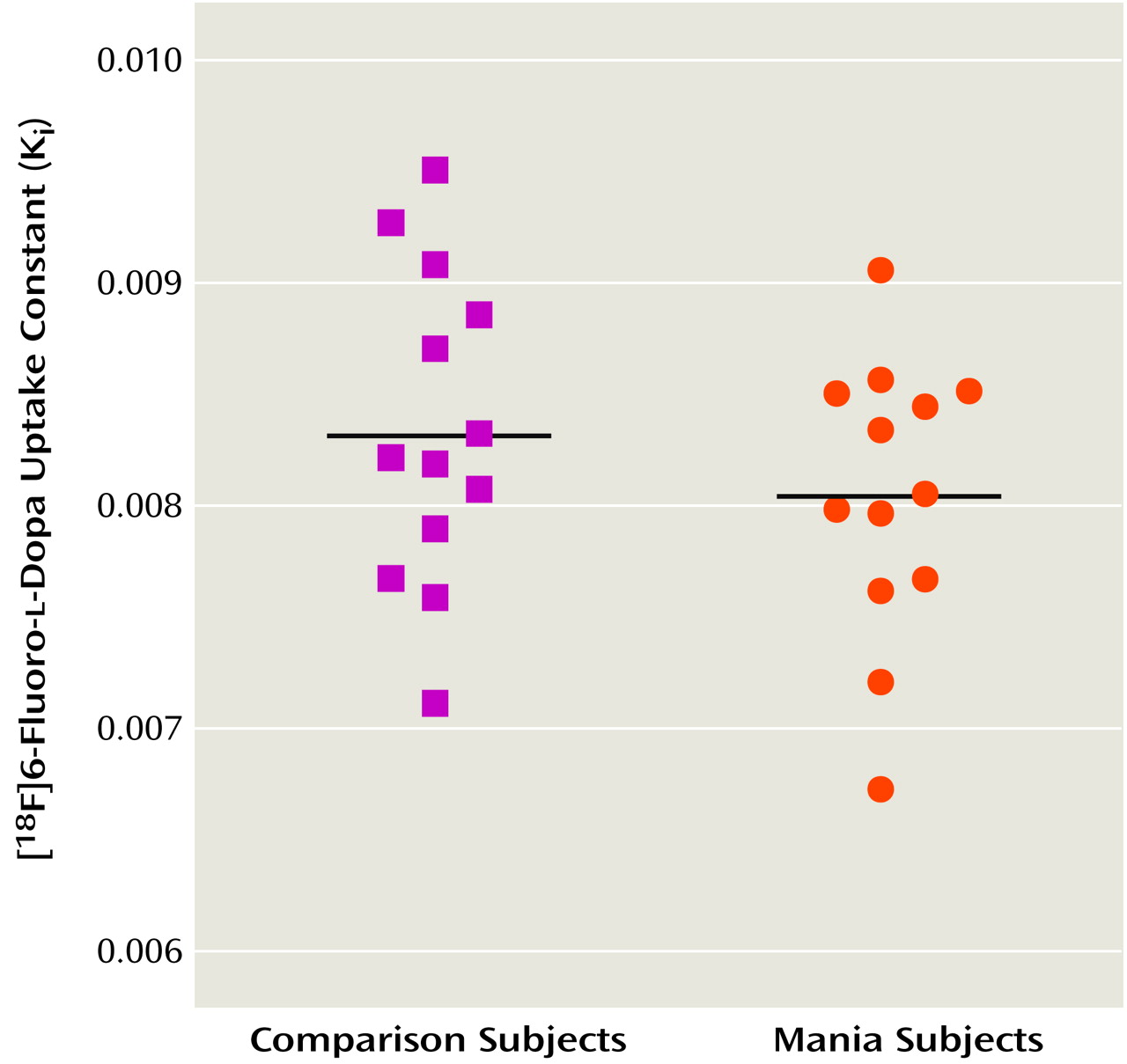
Figure 1 shows scatterplots of K
i values for the striatum in the patients and the comparison subjects. The mean K
i value was 8.04 × 10
–3 (SD=0.62 × 10
–3) for the patients and 8.34 × 10
–3 (SD=0.70 × 10
–3) for the comparison subjects. The difference in K
i values between the two groups was not significant (t=1.13, df=24, p=0.27). Further exploratory analysis comparing K
i values in the caudate, putamen, and right and left striatum also showed no significant differences between patients and comparison subjects (data not shown). There was no significant correlation between patients’ Young Mania Rating Scale scores and pre- or posttreatment K
i values for the striatum.
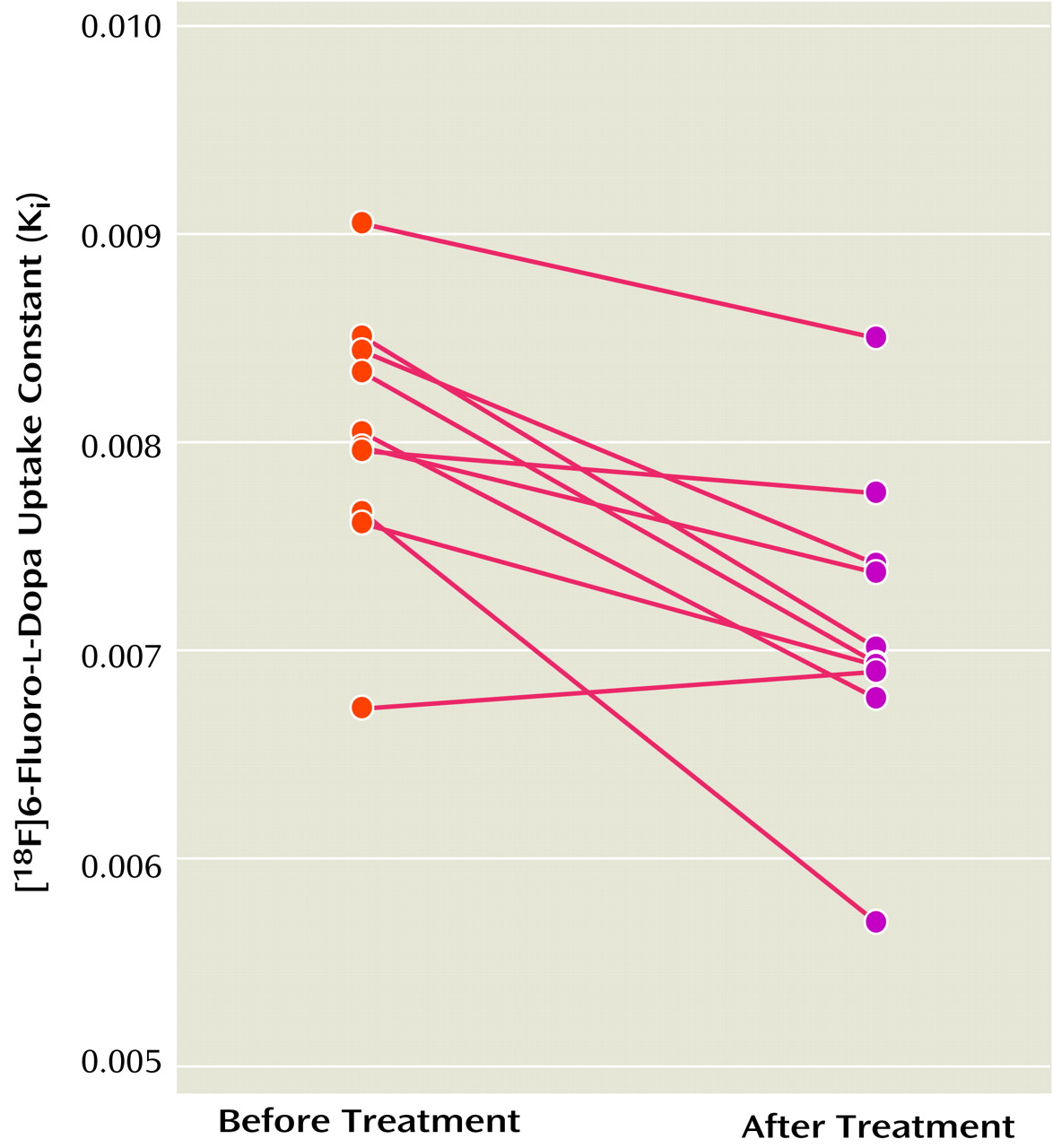
The K
i values for the striatum before and after treatment with divalproex sodium for the 10 patients with complete data are shown in
Figure 2. The mean K
i values decreased by 11.3% after treatment (pretreatment: K
i=8.03 × 10
–3 [SD=0.62 × 10
–3]; posttreatment: K
i=7.12 × 10
–3 [SD=0.72 × 10
–3]), a significant difference (t=4.38, df=9, p=0.002). Further exploratory analysis showed significant decreases with treatment in K
i values for the left striatum (t=4.44, df=9, p=0.002), right striatum (t=2.83, df=9, p=0.02), putamen (t=4.90, df=9, p=0.001), and caudate (t=2.32, df=9, p<0.05). The putamen had the largest decrease in K
i values (i.e., 13%).
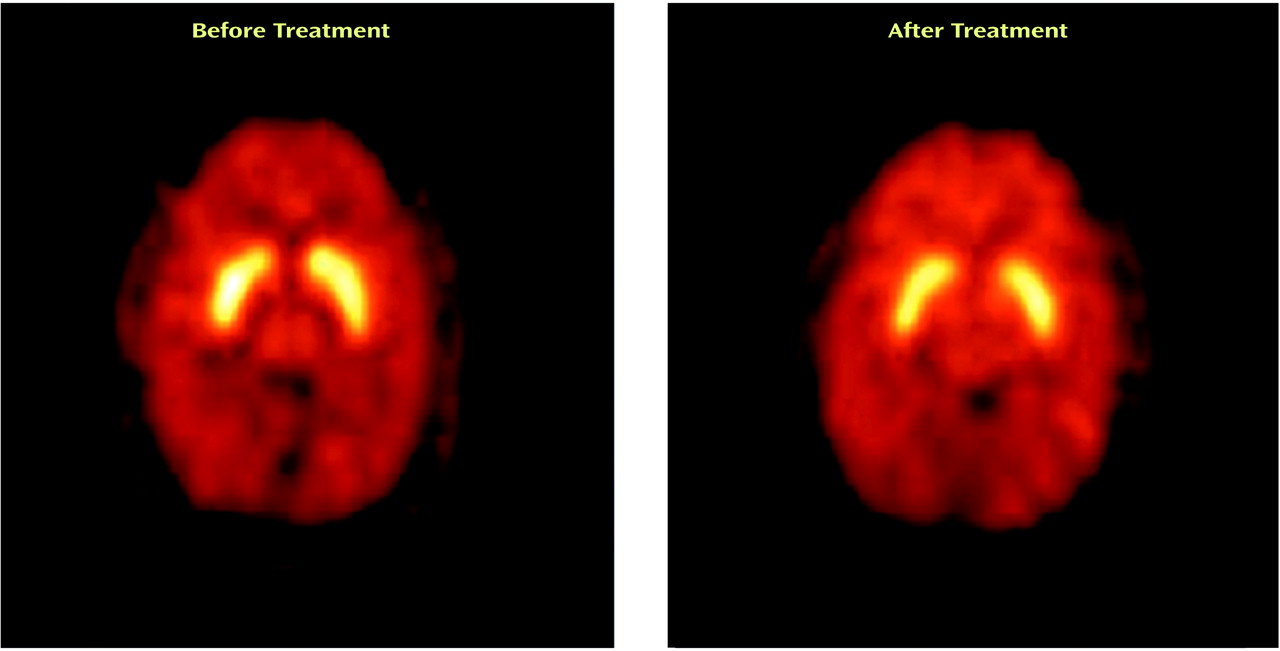
Figure 3 shows PET images displaying activity in the striatum before and after treatment with divalproex sodium.
Patients had significantly lower posttreatment Ki values (about 15% lower) than the healthy comparison subjects (t=–4.03, df=21, p=0.001), suggesting that the presynaptic dopamine function in patients after divalproex sodium treatment was lower than in the comparison subjects. There was no correlation between Young Mania Rating Scale change scores and changes in Ki values from baseline to posttreatment. There was also no correlation between the changes in [18F]DOPA uptake and divalproex sodium dose or duration of treatment.
Discussion
To our knowledge, this is the first study to examine [18F]DOPA uptake in neuroleptic- and mood-stabilizer-naive acutely ill manic patients. The lack of studies involving this group of patients is undoubtedly related to difficulty in recruiting medication-free manic patients for research. Recruitment of 13 medication-free patients for this study took nearly 5 years. The most important findings of this study are that the manic patients showed a significant decrease in [18F]DOPA uptake after treatment with divalproex sodium and that [18F]DOPA uptake in the patients after treatment with divalproex sodium was lower than in the healthy comparison subjects. However, there were no differences in baseline [18F]DOPA uptake between the manic patients and the healthy comparison subjects. There was also no correlation between [18F]DOPA uptake and severity of manic symptoms as measured by Young Mania Rating Scale scores.
The uptake rate constant for [
18F]DOPA in the brain reflects the sum of the blood-brain barrier transport, the decarboxylation by the aromatic
l-amino acid decarboxylase (AADC) to [
18F]dopamine as well as vesicular storage of [
18F]dopamine
(47–
49). Although it is unclear whether [
18F]dopamine synthesis from exogenous [
18F]DOPA reflects endogenous dopamine synthesis, the kinetic constants for 6-fluoro-
l-dopa suggest that this is as good a substrate for AADC as
l-dopa
(50). Furthermore, [
18F]dopamine mimics central dopamine metabolism remarkably well. Therefore, the [
18F]DOPA rate constant should provide an index of AADC activity and transport and storage of [
18F]dopamine into storage vesicles.
The lack of a significant difference in the [
18F]DOPA rate constants between the manic patients and the comparison subjects in this study suggests that this component of presynaptic dopamine function was not increased in the manic patients. Because of the small size of the study group, the possibility of a type II error can not be excluded. Nonetheless, this study had 80% power to detect a 9% difference in [
18F]DOPA rate constants between patients and comparison subjects, and it is unlikely that differences smaller that 9% are clinically meaningful. The mean K
i value for the patients was numerically lower than that for the comparison subjects (i.e., mean difference=–2.96 × 10
–4), but the 95% confidence interval for the mean difference was –8.38 × 10
–4 to 2.44 × 10
–4. The [
18F]DOPA rate constants reflect the sum of activity of AADC as well as transport and storage of [
18F]dopamine into storage vesicles. Thus, our results suggest that these processes are intact in manic patients. However, our findings cannot exclude the possibility of enhanced dopamine release from presynaptic neurons, with consequent increase in dopaminergic transmission in mania. To our knowledge, no study has directly assessed the magnitude of dopamine release in acute mania. However, a recent SPECT study of change in D
2 receptor availability after amphetamine challenge in euthymic bipolar disorder patients failed to demonstrate enhanced dopamine release
(39). This study nevertheless revealed a higher level of behavioral responses (i.e., manic/hypomanic-like symptoms) to amphetamine challenge in the patients, compared to the healthy subjects. These results suggest enhanced postsynaptic dopamine receptor sensitivity and/or second messenger signaling pathway activity in euthymic bipolar disorder patients that may have led to induction of manic/hypomanic symptoms.
If enhancing dopamine transmission induces or worsens manic symptoms, it is plausible that effective antimanic treatments dampen dopamine hyperactivity by decreasing dopamine synthesis, release, or blockade of dopamine receptors or by dampening second messenger signaling pathways. Although the manic patients in this study did not have an abnormality in [
18F]DOPA rate constants at baseline, they showed a significant reduction in [
18F]DOPA rate constants after treatment with divalproex sodium. We did not have a comparison group that had the second scan after placebo or no treatment, and the normal volunteers in this study did not have a second scan. Therefore, one could argue that the reduction in K
i values in the patients could be due simply to scan-to-scan variation in these values and not to a treatment effect. However, the decrease in K
i values observed in the patients was greater than the scan-to-scan variation in K
i values found in normal healthy subjects in a previous study
(43). Furthermore, the decrease in K
i values occurred in nine of the 10 patients. In addition, the decrease occurred in all areas of the striatum, but the largest decrease was noted in the putamen. Also, [
18F]DOPA rate constants were lower in patients after divalproex sodium treatment than in the healthy comparison subjects. Taken together, these findings suggest that the reduction in [
18F]DOPA rate constants was due to the effect of treatment with divalproex sodium or was a correlate of symptomatic change in the manic patients.
We did not find a correlation between changes in Young Mania Rating Scale scores and changes in Ki values between baseline and posttreatment scans in the patients. This result may suggest that the decrease in [18F]DOPA rate constants in the patients after divalproex sodium treatment may not be related to symptom change per se but instead may represent a nonspecific effect of divalproex sodium. On the other hand, the negative finding could be due to the fact that we did not separately measure [18F]DOPA uptake in the ventral striatum (due to the limited resolution of the PET camera). The separate measurement may have correlated better with symptom change (because mesolimbic dopamine transmission is more likely related to symptom change) than the sum of [18F]DOPA uptake in the dorsal and in the ventral striatum.
The results of this study cannot tell us whether the reduction in [
18F]DOPA rate constants after treatment in the manic patients was due to a decrease in AADC activity or interference with the vesicular monoamine transporter 2 (VMAT2) site that mediates transport of dopamine from cytoplasm to storage vesicles. To our knowledge, no previous studies have directly assessed the effects of divalproex sodium on AADC activity or VMAT2 concentrations either in animals or in humans. However, previous evidence suggests that VMAT2 concentrations are not modulated by short- or long-term administration of drugs that affect monoamine function or metabolism
(51). Furthermore, a recent SPECT study reported no alteration in VMAT2 concentrations, as measured by carbon 11-labeled dihydrotetrabenazine binding, in the caudate nuclei of euthymic bipolar disorder patients
(40). Hence, the reduction in [
18F]DOPA rate constants after treatment with divalproex sodium in the current study was more likely to be due to a decrease in AADC activity.
Decarboxylation of DOPA to dopamine by AADC is not rate limiting under the physiological conditions of dopamine synthesis. However, AADC is the catalyst for the first irreversibly committed step in the dopamine synthesis pathway and hence plays an important role in dopamine homeostasis
(52). Indeed, studies have shown that AADC activity is enhanced by dopamine denervation or by dopamine D
2 and D
1 antagonists and is reduced by D
2 agonists
(53,
54). Similarly, AADC is regulated by endogenous dopamine levels, as depletion of dopamine by reserpine was shown to enhance AADC activity
(53). Therefore, reduction in AADC activity with divalproex sodium treatment in this study would be expected to result in a reduction in dopamine synthesis, leading to a lower amount available for release and consequent reduction in dopamine hyperactivity.
The mechanism by which divalproex sodium reduces AADC activity is unknown. The divalproex sodium may alter the activity of AADC through pre- or posttranslational regulation. Administration of valproate for up to 32 days had no effect on mRNA levels of AADC in the rat
(55). AADC protein contains a number of recognition motifs for phosphorylation by cAMP-dependent kinase, protein kinase C, calcium calmodulin-dependent protein kinase II, and proline-directed protein kinase
(56), and it is possible that divalproex sodium may decrease the activity of AADC through inhibition of phosphorylation. However, this possibility needs to be tested directly in future studies.
Neuroleptics—such as the three standard mood stabilizers lithium, valproate, and carbamazepine—have proven antimanic properties. It is widely recognized that dopamine D
2 receptor blockade contributes to the antimanic properties of neuroleptics, whereas the mechanism (or mechanisms) by which mood stabilizers exert an antimanic effect is currently unknown. Recent theories postulate that the effects of these drugs on second messenger signaling pathways are critical to the mood stabilizing properties, although to our knowledge, there is no direct evidence linking the two. Waldmeier
(57), surveying the effects of mood stabilizers on various neurotransmitter pathways, concluded that the effects of these drugs are most similar with respect to reducing dopaminergic transmission, suggesting that this action may represent a common basis for the antimanic effect of these drugs. Reduction in AADC activity in manic patients after treatment with divalproex sodium in this study is consistent with such hypothesis.
In conclusion, the results of this study suggest no difference between acutely ill manic patients and healthy comparison subjects in the [18F]DOPA rate constant that provides an index of a component of presynaptic dopamine function. Treatment with divalproex sodium, however, led to a significant reduction in presynaptic dopamine function in manic patients, suggesting that reduction in AADC activity is associated with improvement in manic symptoms.