For many patients, the effectiveness of current treatments for bipolar disorder is suboptimal. Although lithium, divalproex sodium, and olanzapine—the approved treatments for acute mania—show similar efficacy, clinical trials report nonresponse rates of up to 50% with these agents
(1).
Antipsychotics are commonly used for the initial treatment of acute mania, both as monotherapy and as adjuncts to mood stabilizers
(1). In acute mania, typical antipsychotics show similar efficacy to lithium
(2), but they may have a faster onset of action
(3) and they appear more effective in treating psychomotor agitation
(4,
5). In addition to olanzapine, more recent clinical studies have demonstrated the efficacy of other atypical antipsychotics in treating acute mania at doses effective for schizophrenia
(2). Generally, atypical antipsychotics have a lower risk of neurological side effects compared with typical agents and may be less likely to exacerbate depressive symptoms
(6). However, currently available atypical antipsychotics are associated with unwanted side effects, including weight gain
(7), hyperprolactinemia
(8), QTc prolongation
(9), hyperglycemia
(10,
11), and dyslipidemia
(12).
Aripiprazole is a novel antipsychotic with a pharmacodynamic profile that distinguishes it from other antipsychotics. Aripiprazole is a partial agonist at dopamine D
2 receptors
(13,
14), a partial agonist at serotonin 5-HT
1A receptors
(15), and an antagonist at 5-HT
2A receptors
(16). In randomized, placebo-controlled studies in patients who experienced an acute relapse of schizophrenia or schizoaffective disorder, aripiprazole improved positive and negative symptoms and demonstrated a favorable safety and tolerability profile, with low liability for extrapyramidal symptoms, weight gain, sedation, hyperprolactinemia, or QTc prolongation
(17).
The objective of the current study was to assess the efficacy, safety, and tolerability of aripiprazole for the treatment of patients in a manic or mixed episode of bipolar I disorder.
Method
Patients
This study enrolled male and female patients, age ≥18 years, diagnosed with bipolar I disorder, manic or mixed episode (DSM-IV), who were experiencing an acute relapse that required hospitalization. Female patients of child-bearing potential were required to agree to use acceptable contraceptive measures and were excluded if they were lactating or pregnant. In addition, patients were required to have a Young Mania Rating Scale
(18) score of ≥20 at the time of randomization.
Patients were excluded from the study if they had delirium, dementia, amnestic or other cognitive disorders, schizophrenia, or schizoaffective disorder or if they were experiencing their first manic episode. Other exclusion criteria were duration of current mania >4 weeks; nonresponse to clozapine; probable need for prohibited concomitant therapy (neuroleptic agents, fluoxetine, carbamazepine, divalproex sodium, valproic acid or sodium valproate, lithium, benzodiazepines [except lorazepam], and all other psychotropic drugs) during the study; use of psychoactive substances or a substance use disorder; serum concentrations of lithium >0.6 mmol/liter or divalproex sodium >50 μg/ml (i.e., therapeutic levels) at screening; significant risk of committing suicide or homicide; history of neuroleptic malignant syndrome or seizure disorder; clinically significant abnormal laboratory test results, vital signs, or ECG results; or previous enrollment in an aripiprazole clinical trial.
After complete description of the study to the subjects, written informed consent was obtained from them or from a legally acceptable representative if required by the institutional review board or ethics committee for their study center. The study protocol, procedures, and consent statement were approved by the institutional review boards of each participating site.
Study Design
This was a multicenter, double-blind, randomized, placebo-controlled study. After a 1–7 day screening period to assess patient eligibility and allow elimination of existing psychotropic medications, patients fulfilling study entry criteria were randomly assigned to receive aripiprazole, 30 mg/day, or placebo. During the study, the aripiprazole dose could be reduced to 15 mg/day for tolerability if needed.
The treatment period lasted 3 weeks. Patients were hospitalized for at least the first 2 weeks of treatment. At the end of week 2, patients could be discharged if they had the following scores on the Clinical Global Impression (CGI)—Bipolar Version
(19): severity of illness (mania) score ≥3 (mildly ill, minimally ill, or not ill) and a change from preceding phase (mania) score ≥2 (much improved or very much improved). Patients not meeting these criteria stayed in the hospital for the remaining week of treatment. Patients not responding at the end of week 2 (change from preceding phase [mania] score of 4 [no improvement] to 7 [very much worse]) were discontinued from double-blind treatment and offered the option of open-label aripiprazole treatment during week 3.
Efficacy Assessments
Treatment efficacy was assessed at baseline and at days 4, 7, 10, 14, and 21. The primary efficacy measure was mean change in total score (last observation carried forward) on the Young Mania Rating Scale from baseline to the end of study (day 21). The Young Mania Rating Scale consists of 11 items that assess the core symptoms of mania: 1) elevated mood, 2) increased motor activity/energy, 3) sexual interest, 4) sleep, 5) irritability, 6) speech (rate and amount), 7) language/thought disorder, 8) content, 9) disruptive/aggressive behavior, 10) appearance, and 11) insight. The severity of four items (items 5, 6, 8, and 9) is graded from 0 (best) to 8 (worst), while the other seven items are graded from 0 (best) to 4 (worst). The Young Mania Rating Scale total score is the sum of all the items; possible scores range from 0 to 60.
Other efficacy measures included mean change for each of the CGI—Bipolar Version scores for severity of illness and change from preceding phase (i.e., for mania, depression, and overall bipolar illness); discontinuation due to lack of efficacy or entry into open-label aripiprazole treatment; and response (≥50% decrease in Young Mania Rating Scale score from baseline).
Safety and Tolerability Assessments
Reports of adverse events were obtained from patients throughout the study period (including the screening phase). Investigators evaluated reported events for severity and likely relationship to study medication. Patients’ vital signs were measured at screening, baseline, and each assessment visit during the study. Twelve-lead ECGs, blood and urine collection, and body weight measurements were performed at screening and the end of the study.
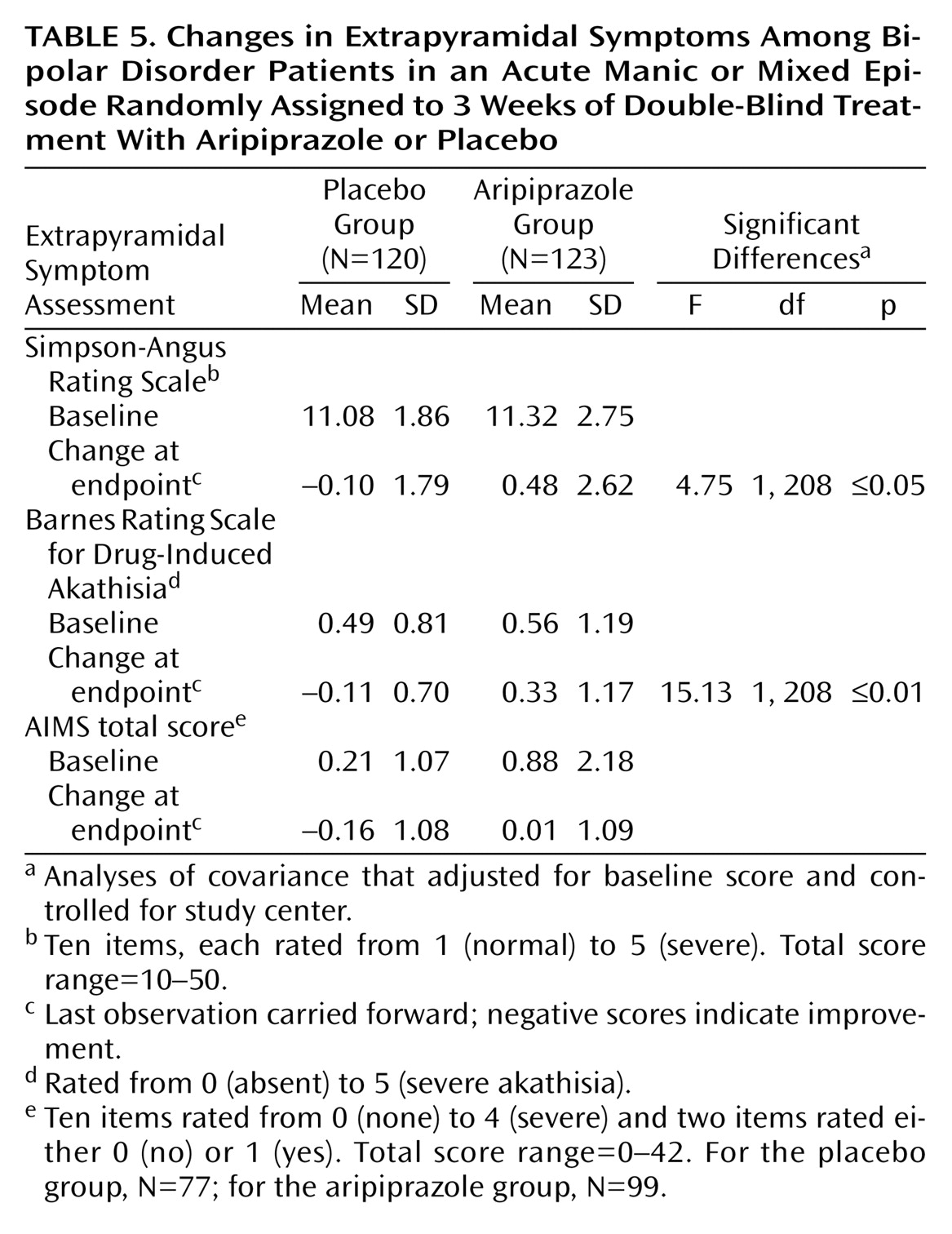
Extrapyramidal symptoms were evaluated by using the Simpson-Angus Rating Scale
(20), the Barnes Rating Scale for Drug-Induced Akathisia
(21), and the Abnormal Involuntary Movement Scale (AIMS)
(22). Evaluations were made at baseline and at each assessment visit for the Simpson-Angus Rating Scale and Barnes Akathisia Scale and at baseline and at the end of the study for the AIMS.
Concomitant Medication
Concomitant medication use was recorded on case report forms. Concomitant use of psychotropic medications (apart from study medication) was prohibited during the study (including the screening phase) except for lorazepam. Lorazepam treatment was allowed only on days 1–4 (≤6 mg/day), 5–7 (≤4 mg/day), and 8–10 (≤2 mg/day).
Anticholinergic treatments for extrapyramidal symptoms were not permitted during screening. Anticholinergic agents were permitted after treatment assignment if deemed necessary by the investigator, but use was limited to 6 mg/day of benztropine (or equivalent) and could not be administered within 12 hours of an efficacy or safety assessment.
Statistical Procedures
Efficacy parameters were analyzed on an intent-to-treat basis from data obtained at each patient’s final visit (i.e., last-observation-carried-forward analysis). Efficacy measures were evaluated by analyses of covariance (ANCOVA) that adjusted for baseline score and controlled for study center; rates of discontinuation due to lack of efficacy or entry into open-label aripiprazole treatment, response rates, and change from preceding phase scores were evaluated by the Cochran-Mantel-Haenszel test, which controlled for study center. Data from small centers, defined as no patients in one or more treatment groups, were pooled to form pseudocenters before analysis.
Simpson-Angus Rating Scale, AIMS, and Barnes Rating Scale for Drug-Induced Akathisia scores were analyzed on an intent-to-treat basis from data obtained at each patient’s final visit by using ANCOVA after we adjusted for baseline score and controlled for study center.
Results
Patients
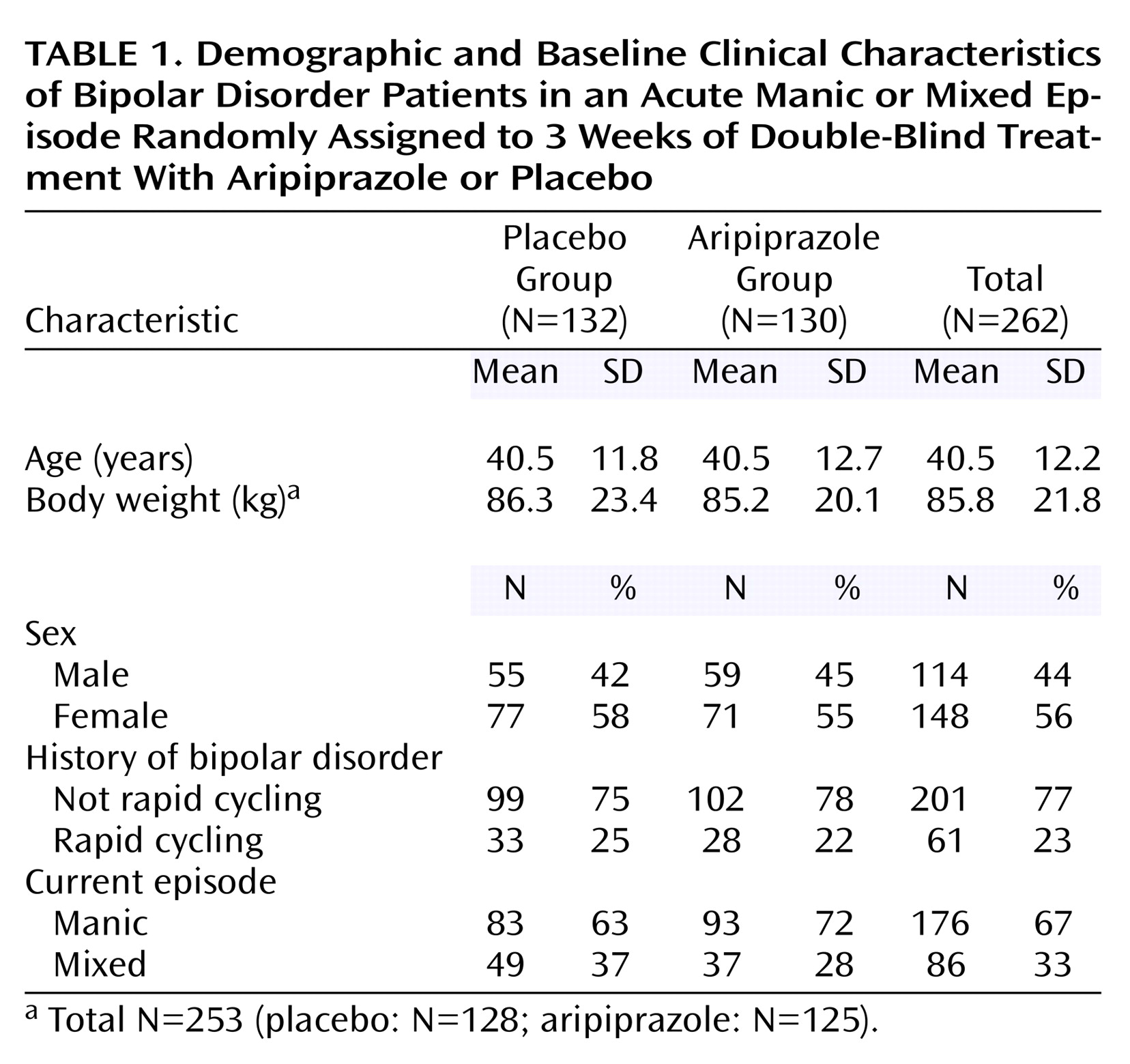
The study was conducted at 38 centers in the United States. Overall, 262 patients were randomly assigned to a study treatment condition (130 to aripiprazole, 132 to placebo); this study is one of the largest double-blind, placebo-controlled trials of an antipsychotic as monotherapy for acute mania reported to date. Baseline characteristics for the patients are shown in
Table 1.
Of the 262 patients assigned to a treatment condition, eight did not take study medication and thus were excluded from safety and efficacy analyses (aripiprazole: N=3, placebo: N=5). An additional six patients (aripiprazole: N=2, placebo: N=4) did not have a valid postrandomization efficacy evaluation because of early discontinuation from the study and so were excluded from the efficacy analysis. Hence, valid data from 254 patients and 248 patients were available for the safety analysis and the efficacy analysis, respectively.
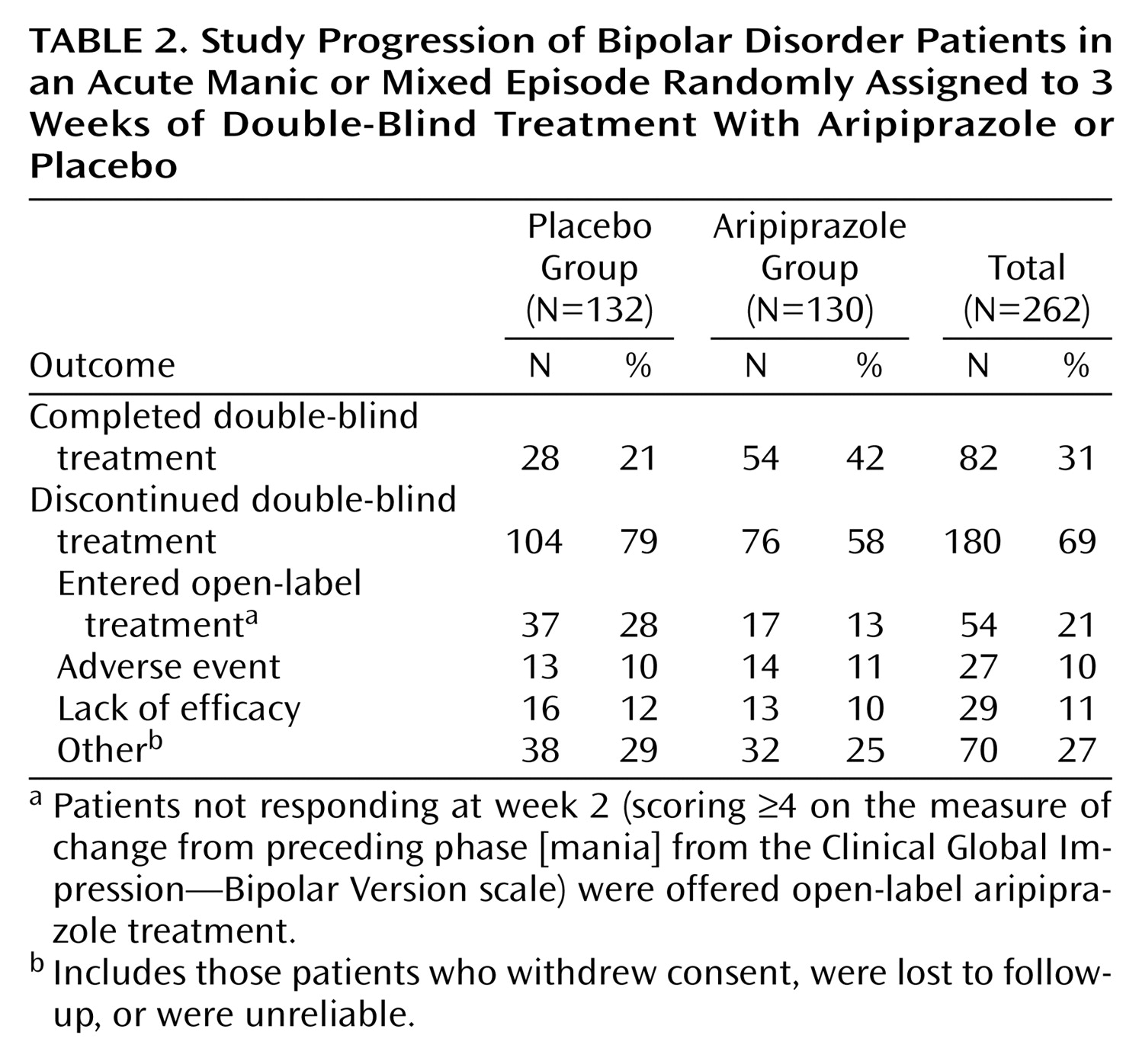
Overall, 82 (31%) patients completed the 3-week, double-blind treatment period. The completion rate was significantly higher with aripiprazole treatment than placebo (42% versus 21%) (χ
2=10.90, df=1, p<0.001). Reasons for discontinuation are shown in
Table 2. The main difference between the two groups was in the number of patients who did not display an adequate response at week 2 and thus entered open-label treatment with aripiprazole. Discontinuations due to adverse events were similar between the two groups.
The mean dose of aripiprazole at endpoint was 27.9 mg/day for patients in the safety analysis. The majority of patients (86%) receiving aripiprazole during double-blind treatment remained on the 30-mg dose throughout the study. The number of patients requiring lorazepam treatment during the study was comparable in each treatment group (placebo: 108 of 127 [85%]; aripiprazole: 109 of 127 [86%]).
Efficacy
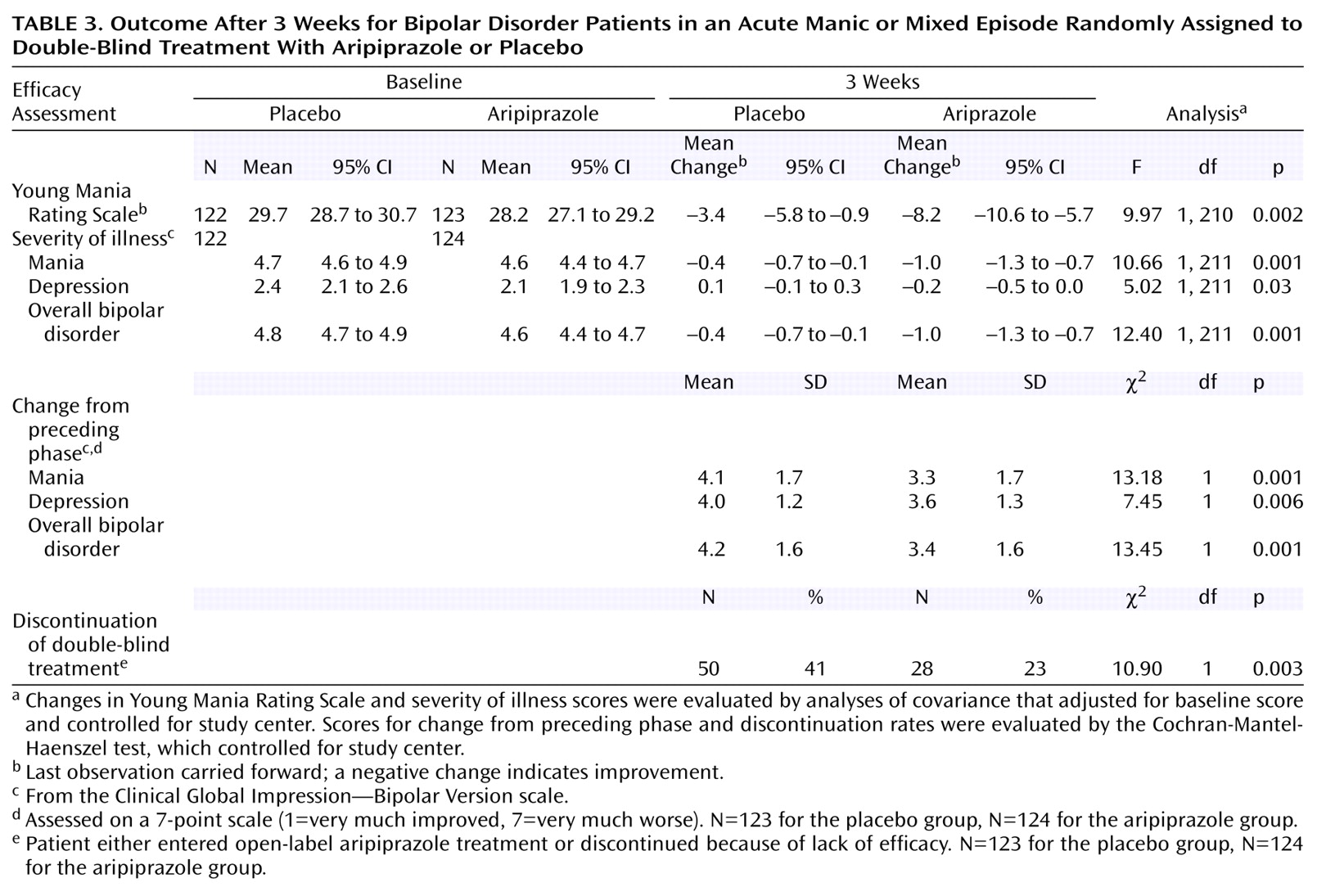
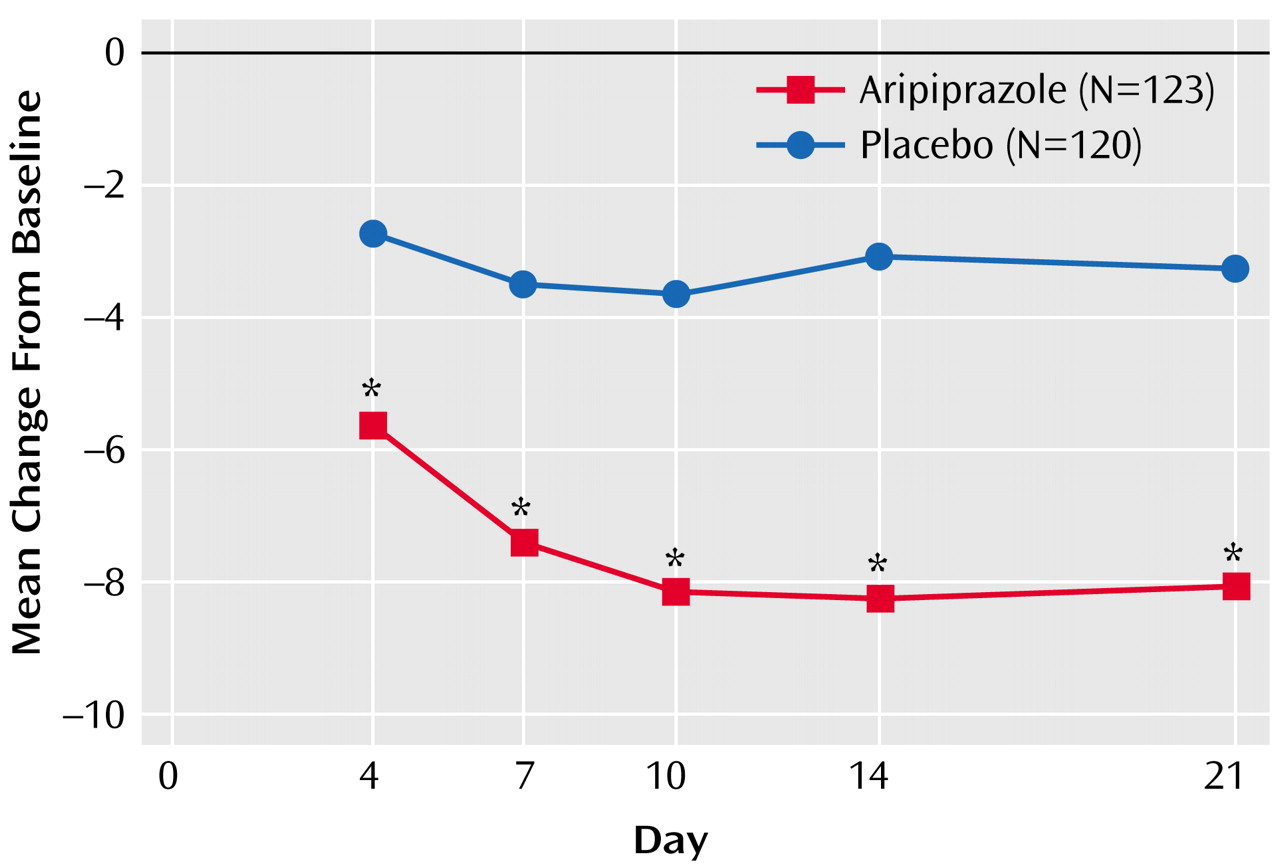
Aripiprazole produced a significant improvement in total scores on the Young Mania Rating Scale from baseline to endpoint compared with placebo (
Table 3). The significant difference in mean change between the aripiprazole and placebo group was apparent from day 4 onward (
Figure 1).
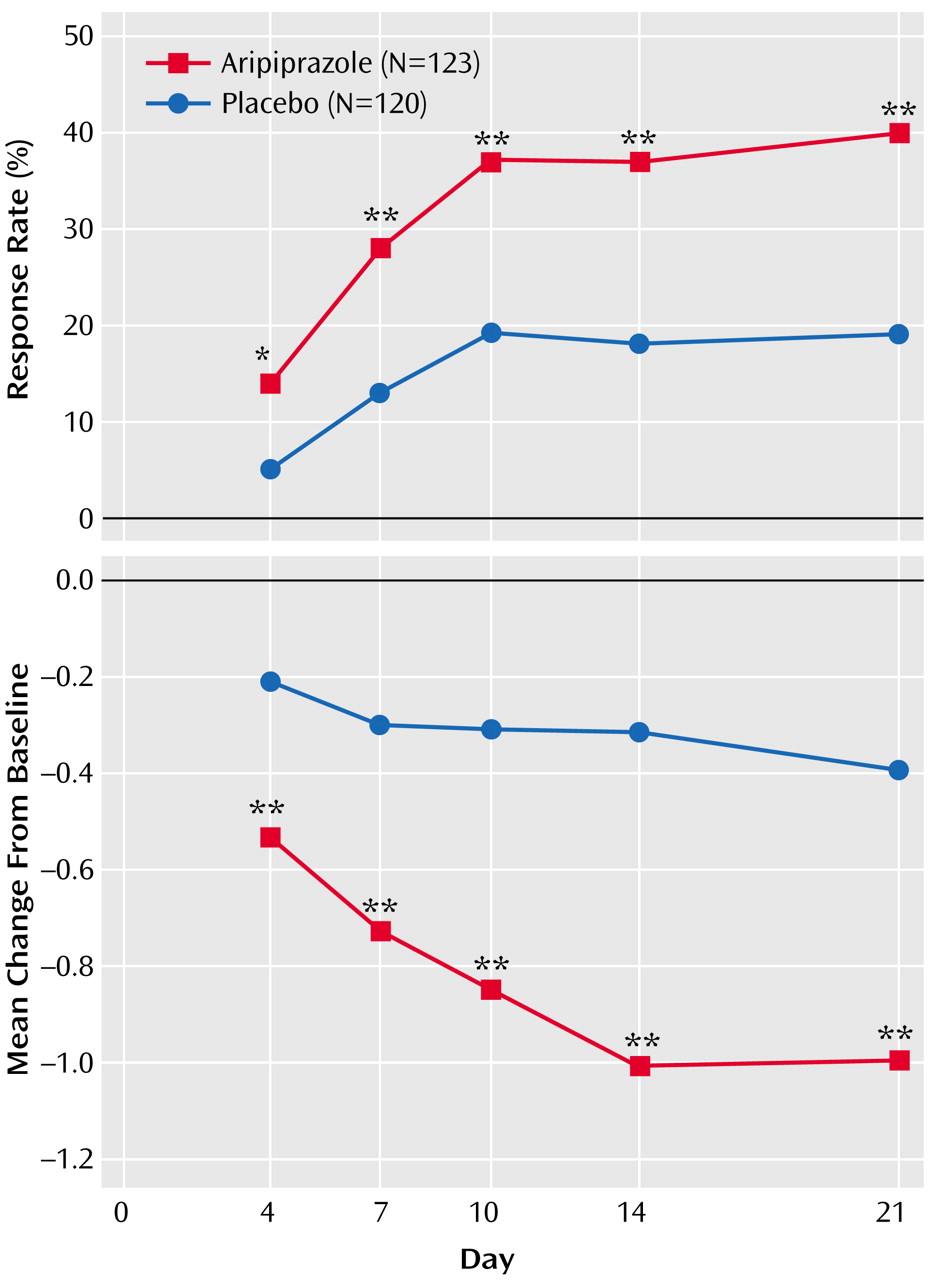
Response rates were significantly greater with aripiprazole than placebo at all time points from day 4 (14% versus 5%) through endpoint (40% versus 19%) (
Figure 2). Aripiprazole produced significantly greater improvements in severity of illness (mania) scores compared with placebo (
Table 3), which were seen at day 4 and all subsequent efficacy assessments (
Figure 2). Aripiprazole also produced significantly superior mean scores from day 4 onward compared with placebo for the change from preceding phase (mania) score (
Table 3).
In addition to the mania ratings, aripiprazole also produced significantly greater improvements for the CGI—Bipolar Version severity of illness scores for depression and overall bipolar illness and significantly superior change from preceding phase scores for depression and overall bipolar illness than were seen with placebo (
Table 3).
Safety
Adverse events
In total, 27 (11%) of the 254 patients available for the safety analysis discontinued from double-blind treatment because of an adverse event, 13 from the placebo group and 14 from the aripiprazole group.
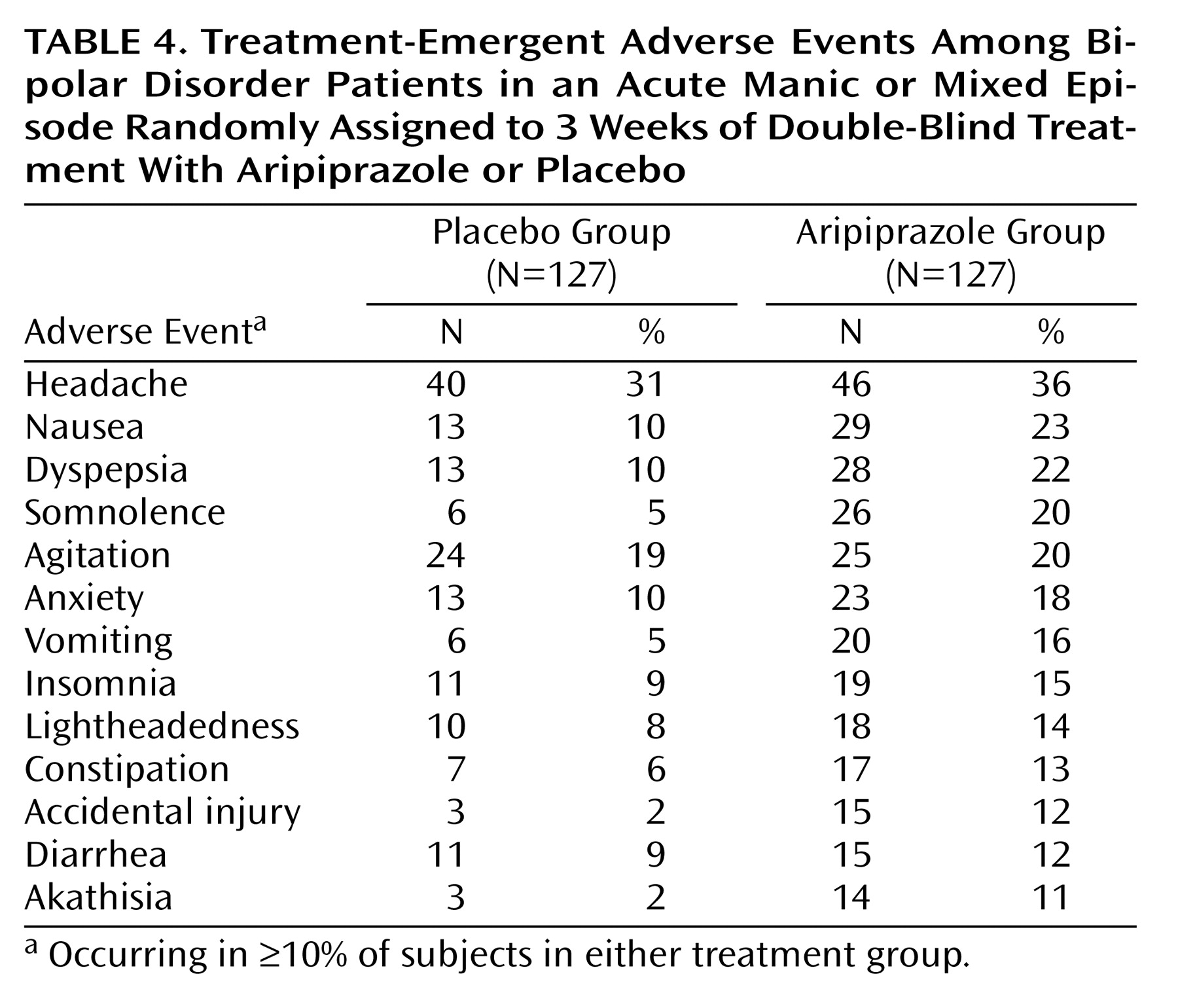
Overall, 11 serious adverse events were experienced by eight patients (3%), four from each treatment group, during double-blind treatment or within 30 days of discontinuation (one patient receiving placebo reported two serious adverse events, and one patient receiving aripiprazole reported three serious adverse events). The reported events in the aripiprazole group were manic reaction (N=3), psychiatric decompensation, overdose of sedatives, and hypertension (one event each). The placebo group reported agitation, accidental injury, chest discomfort, syncope, and urticaria (one event each). The most common, treatment-emergent adverse events are shown in
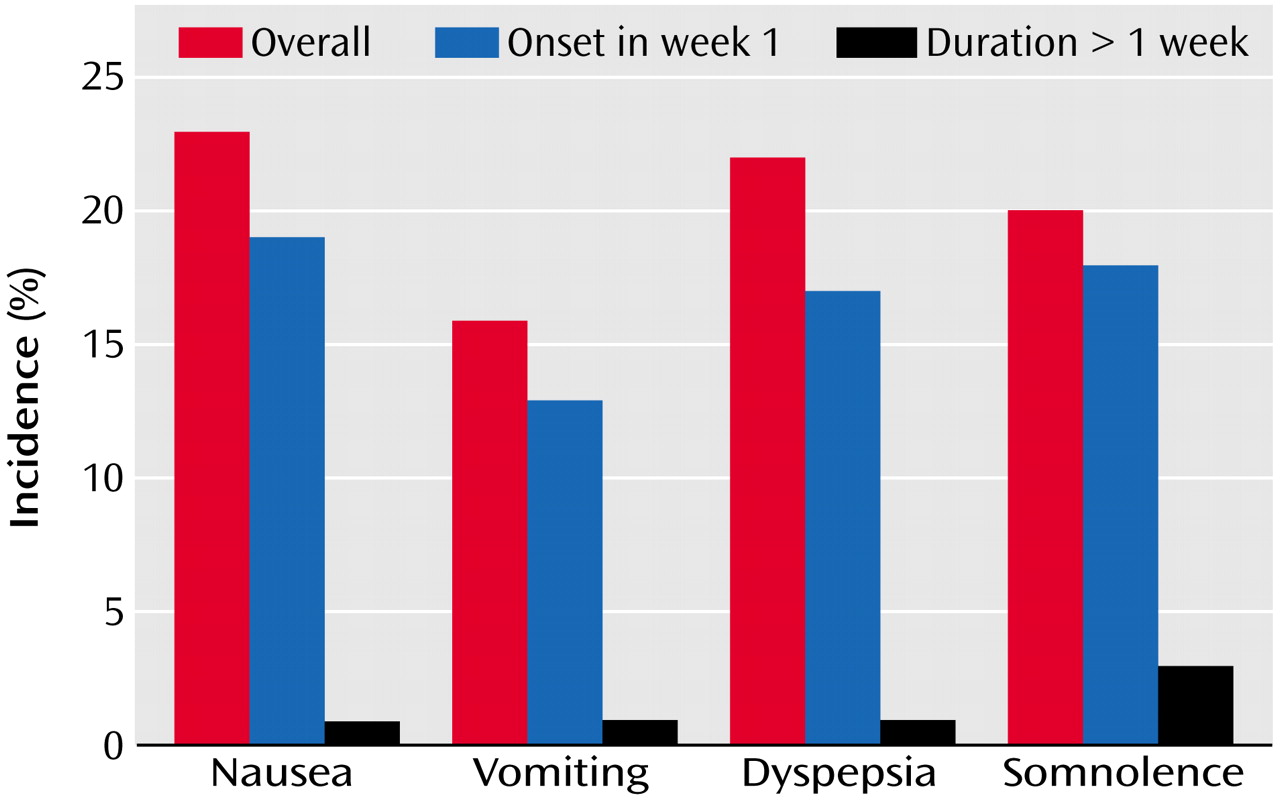
Table 4. Further analysis of somnolence and gastrointestinal events showed that the majority of events occurred during the first week of treatment and resolved within 7 days (
Figure 3).
Extrapyramidal symptoms
The most commonly reported extrapyramidal symptom-related adverse events were akathisia (11%) and tremor (6%) with aripiprazole and tremor and extrapyramidal syndrome (both 3%) with placebo. Both dose reduction (3%) and discontinuation due to akathisia (1.6%) were infrequent in patients receiving aripiprazole.
Changes from baseline for Simpson-Angus Rating Scale and Barnes Rating Scale for Drug-Induced Akathisia scores were small but significantly greater in the aripiprazole group compared with the placebo group. Changes from baseline in AIMS score did not differ significantly between the groups (
Table 5).
Body weight
Patients in both treatment groups experienced a slight decrease in mean body weight from baseline to endpoint (placebo: –0.8 kg; aripiprazole: –0.3 kg). No statistically significant difference in clinically significant weight gain (≥7% increase from baseline) was detected between the two groups (aripiprazole, N=2; placebo, N=0).
Prolactin
Mean baseline prolactin values were within normal limits for male patients (15.5 ng/ml) and female patients (25.7 ng/ml) in the placebo group and for male patients (18.1 ng/ml) in the aripiprazole group but were above the upper limit of normal for female patients (31.0 ng/ml) in the aripiprazole group. Over the course of the study, mean serum prolactin levels decreased by 12.7 ng/ml (SD=29.0) in the aripiprazole group and 7.2 ng/ml (SD=28.1) in the placebo group (F=6.53, df=1, 175, p≤0.05). Mean endpoint values for both treatment groups, 14.1 ng/ml and 14.8 ng/ml, respectively, were within normal limits.
The percentage of patients with a serum prolactin value above the upper limit of normal during study treatment was higher in the placebo group (17%) than the aripiprazole group (11%). At baseline, 23% of patients in the placebo group and 30% of those in the aripiprazole group had prolactin values above the upper limit of normal.
ECG
One patient who received placebo experienced a clinically significant increase in QTc interval (QTc ≥450 msec and a ≥10% increase from baseline) when Bazett’s correction factor was applied. This was normalized when the Food and Drug Administration Neuropharmacological Division correction factor was used. No patients receiving aripiprazole were determined to have a clinically significant increase in QTc interval.
Vital Signs and Laboratory Analyses
There were no clinically significant differences between groups in vital sign changes or laboratory analyses. No patients receiving aripiprazole discontinued from the study due to vital sign or laboratory abnormalities. The rates of patients with clinically significant levels of fasting serum glucose (≥110 mg/dl) or total cholesterol (≥200 mg/dl) were similar in both groups.
Discussion
The results of the current study suggest that aripiprazole was efficacious, safe, and well tolerated for patients with bipolar I disorder experiencing an acute manic or mixed episode. Aripiprazole produced a statistically significant mean decrease in Young Mania Rating Scale total score by day 4 that was maintained throughout the 3-week study period. Twice as many patients in the aripiprazole group responded to treatment as in the placebo group (40% versus 19%); superior response rates with aripiprazole were evident by day 4.
Other outcome measures provide further support for the efficacy of aripiprazole in treating acute mania. Significantly fewer patients from the aripiprazole group discontinued treatment because of lack of efficacy or entry into open-label aripiprazole treatment than did patients in the placebo group. Aripiprazole significantly improved all CGI—Bipolar Version severity of illness and change from preceding phase scores (i.e., scores for depression and overall bipolar illness as well as scores for mania). The global improvement in depressive symptoms is important in the context of concerns regarding the depressogenic effects of typical antipsychotics in bipolar disorder
(6).
Significantly more patients in the aripiprazole group (N=54 [42%]) completed double-blind treatment than in the placebo group (N=28 [21%]). However, forced discontinuation of study drug was a prespecified criterion in patients not responding at week 2 (scoring ≥4 on the measure of change from preceding phase [mania]), and patients were given the option of entering into an open-label aripiprazole treatment group for week 3. Thirteen patients randomly assigned to double-blind treatment with aripiprazole entered and completed open-label treatment, accounting for an overall completion rate of 67 (52%) in aripiprazole-treated patients. This completion rate is comparable to those seen in two placebo-controlled studies with olanzapine in mania
(23,
24).
The primary reason leading to discontinuation in the study was withdrawal of consent, which accounted for 22% of discontinuation in the aripiprazole group and 23% with placebo. The discontinuation rate due to lack of efficacy or adverse events with aripiprazole treatment in the current study (22%) was comparable to the rates of 29% and 31% reported with olanzapine in placebo-controlled studies
(23,
24) and to that of a 3-week study with ziprasidone (26%)
(25).
Aripiprazole treatment was well tolerated during the study. Adverse events were generally mild to moderate and tended not to be treatment-limiting. The majority of patients (86%) remained on the 30-mg/day dose during the study. The rate of discontinuations due to adverse events was 10% and 11% in placebo and aripiprazole treatment groups, respectively. The incidence of adverse events was similar in both groups, except nausea, dyspepsia, vomiting, constipation, somnolence, accidental injury, and akathisia, which occurred at an incidence of ≥10% in the aripiprazole group and more than twice as frequently than with placebo. Further analysis of somnolence and gastrointestinal side effects revealed that most of these events occurred during the first week of treatment and were of limited duration.
Although aripiprazole was not associated with a significantly different change from baseline AIMS score compared with placebo, there were small but significant changes from baseline in Simpson-Angus Rating Scale and Barnes Rating Scale for Drug-Induced Akathisia scores. The majority of extrapyramidal symptom-related adverse events were mild to moderate in severity. Dose reductions for akathisia (3%) and discontinuation of study medication due to akathisia (1.6%) were both infrequent in the aripiprazole group. Extrapyramidal syndrome and tremor were the only other extrapyramidal symptom-related adverse events that led to discontinuation of study treatment (each, 0.8% of aripiprazole patients).
Weight gain is a frequent problem in patients receiving pharmacological treatment for bipolar disorder
(23,
24,
26–28). Two recent studies concluded that obesity was more prevalent in patients with bipolar disorder than the general population
(29,
30). In the current study, both treatment groups experienced a small decrease in body weight during the study, and there was no significant difference between groups in the incidence of clinically significant weight gain. Mean serum prolactin levels decreased significantly more in the aripiprazole group than with placebo, and none of the patients receiving aripiprazole experienced a clinically meaningful change in QTc interval in random ECGs.
In summary, aripiprazole, 30 mg/day, was effective, safe, and well tolerated in patients with bipolar I disorder experiencing an acute manic or mixed episode. Aripiprazole had a rapid onset of action and provided superior efficacy to placebo that was maintained over the 3-week study period. Treatment with aripiprazole was not associated with weight gain, prolactin elevation, or QTc prolongation, consistent with the findings of earlier studies of patients with schizophrenia. Aripiprazole’s favorable safety and tolerability profile may reflect its novel mechanism of action (dopamine-serotonin system stabilizer). Partial agonist activity at dopamine D
2 receptors has been associated with stabilization of the dopamine system (i.e., functional antagonism in hyperdopaminergic states and functional agonism in hypodopaminergic states)
(31–
33) and may therefore be linked to a decreased risk for side effects associated with D
2 antagonism (e.g., extrapyramidal side effects and hyperprolactinemia)
(34). Longer-term studies in patients with bipolar disorder will be necessary to determine whether the short-term benefits observed in the current study are maintained.
Acknowledgments
The Aripiprazole Study Group consists of the following investigators: A. Ari Albala, M.D.; Anne Andorn, M.D.; Bijan Bastani, M.D.; Louise Beckett, M.D.; Charles Bowden, M.D.; Steven Brannan, M.D.; Ronald Brenner, M.D.; Franca Centorrino, M.D.; Sabah Chammas, M.D.; Christopher Chung, M.D.; Evagelos Coskinas, M.D.; Andrew J. Cutler, M.D.; David Daniel, M.D.; Bradley C. Diner, M.D.; John Downs, M.D.; Eduardo Dunayevich, M.D.; Richard James Farrer, M.D.; Natalie Gershman, M.D.; Donald Hilty, M.D.; Scott Hoopes, M.D.; Robert Lynn Horne, M.D.; Philip G. Janicak, M.D.; Anita Kablinger, M.D.; Terence Ketter, M.D.; Michael Levy, M.D.; Paul Markovitz, M.D., Ph.D.; Howard Keith Mason, M.D.; Denis Mee-Lee, M.D.; Dennis M. Pavlinac, M.D.; Joachim Raese, M.D.; Barry R. Rittberg, M.D.; James Russell, M.D.; Frederick Schaerf, M.D.; Robert Taylor Segraves, M.D., Ph.D.; Anantha Shekhar, M.D.; Raj Shiwach, M.D.; Patricia Suppes, M.D.; Tram K. Tran-Johnson, Pharm.D., Psy.D.; Cherian Verghese, M.D.; Richard H. Weisler, M.D.; Andrew Winokur, M.D.