Depression remains a persistent source of morbidity and mortality for patients with bipolar disorder. Controlled trials suggest that standard antidepressants may offer little therapeutic advantage beyond the effects of therapeutically dosed mood stabilizers
(1,
2), and all appear to heighten risk for inducing mania or cycle acceleration
(3).
Dopamine agonists have gained increasing attention for their possible antidepressant effects. Preclinical evidence of hypodopaminergic tone in depression derives from reduced homovanillic acid levels and increased mesolimbic function after tricyclic antidepressant therapy
(4). Monoamine oxidase inhibitors may be advantageous for anergic depressions, partly because of their prodopaminergic effects
(5). Pramipexole is a novel D2/D3 agonist previously shown to exert antidepressant efficacy comparable to that of fluoxetine for major depression
(6). Case reports
(7) and open trials
(8,
9) suggest the utility of pramipexole in bipolar and treatment-resistant depression. The present pilot study was undertaken to provide a clearer estimate of the antidepressant efficacy and safety of pramipexole compared with placebo added to mood stabilizers in outpatients with treatment-resistant bipolar depression.
Method
Subjects for the present study were 22 outpatients with DSM-IV bipolar disorder experiencing major depression defined by DSM-IV criteria. The patients, recruited by advertisement, word of mouth, and clinic referral, were seen in the Bipolar Disorders Research Clinic of the Payne Whitney Clinic, New York Presbyterian Hospital. All had not responded to at least two adequate trials of standard antidepressants with concomitant mood stabilizers during the current episode. We made research diagnoses of bipolar disorder based on the Structured Clinical Interview for DSM-IV. Depressive and manic symptoms were rated by using the 17-item Hamilton Depression Rating Scale
(10) and the Young Mania Rating Scale
(11) after achieving adequate interrater reliability. Clinical status was secondarily assessed by using the Clinical Global Impression (CGI) severity scale
(12). Induction of mania or hypomania was defined as a Young Mania Rating Scale score greater than 15 after study initiation.
At baseline, all patients were nonpsychotic and had Young Mania Rating Scale scores less than 12 and Hamilton depression scale scores greater than 18. None took antipsychotic medications during the study, and all had been taking lithium, divalproex, carbamazepine, lamotrigine, and/or topiramate at stable doses for the month before random assignment to placebo or pramipexole; their doses of these drugs were held constant throughout the study. Concomitant lorazepam (up to 2 mg/day) or clonazepam (up to 1 mg/day) were permitted as needed for insomnia or agitation.
Patients were randomly assigned to placebo or active drug by an unblinded research assistant. Ten patients were assigned to placebo and 12 to active drug. Pramipexole administration was started at 0.125 mg twice a day and increased by 0.25 mg/day every 3–5 days to a target range of 1.0–2.5 mg/day. Higher doses (up to 5.0 mg/day) were permitted as needed. Dose escalations continued until 1) achievement of primary endpoint (defined as a reduction of 50% or more from baseline in Hamilton depression scale score for at least 2 successive weeks), 2) drug intolerance, or 3) 6-week protocol completion.
All patients provided written informed consent to participate in the study protocol, which was approved by the Institutional Review Board of the Weill Medical College of Cornell University-New York Presbyterian Hospital.
Efficacy and safety analyses were conducted on all patients who completed at least 1 week of treatment. Analyses were based on last observations carried forward. Proportions of responders and nonresponders in each condition were compared by chi-square or Fisher’s exact tests. Changes from baseline are reported as differences in means and standard deviations and analyzed with Mann-Whitney tests. All statistical tests were two-tailed with an alpha level of 0.05.
Results
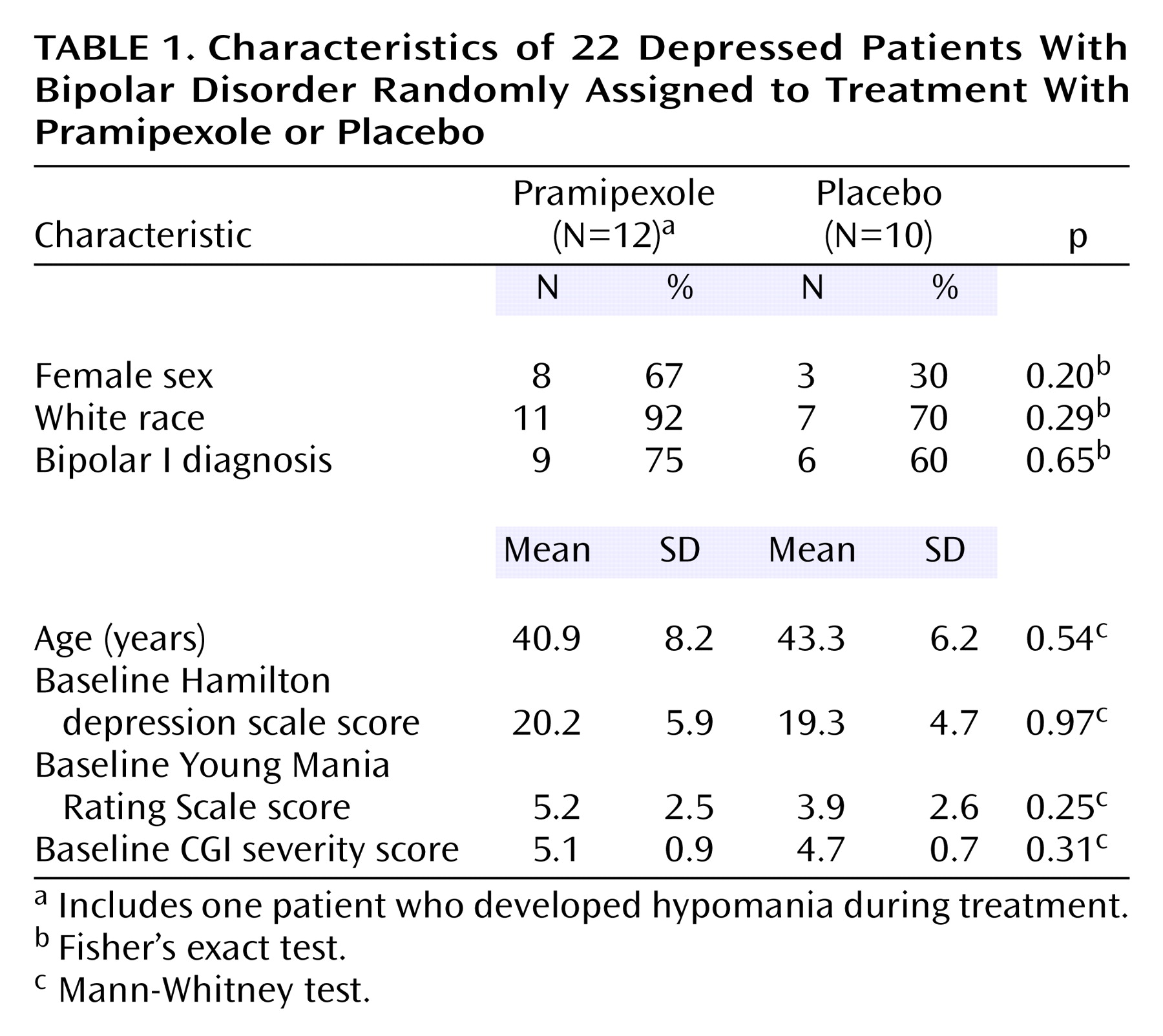
Characteristics of patients taking pramipexole or placebo are summarized in
Table 1. The mean peak dose of pramipexole was 1.7 mg/day (SD=1.3). Six-week study completion rates were somewhat higher for patients taking pramipexole (10 [83%] of 12) than for patients taking placebo (six [60%] of 10) (χ
2=1.50, df=1, p=0.22). Mean concomitant medications, dosed alone or in combinations, included lithium (N=6): mean=1137.5 mg/day (SD=381.6) (serum Li
+ mean=0.70 meq/liter, SD=0.21); divalproex (N=9): mean=916.7 mg/day (SD=129.1) (serum valproate mean=80.7 μg/ml, SD=15.4); carbamazepine (N=2): mean=400.0 mg/day (SD=282.8); lamotrigine (N=6): mean=283.3 mg/day (SD=144.3); and gabapentin (N=3): mean=450 mg/day (SD=212.1).
A reduction of 50% or more from baseline Hamilton depression scale was evident among eight (67%) of 12 patients taking pramipexole and two (20%) of 10 taking placebo (p=0.04, Fisher’s exact test). The mean change from baseline in Hamilton depression scale scores was greater for patients taking pramipexole (mean=48.0%, SD=33.1%) than for those taking placebo (mean=21.4%, SD=36.3%) (p=0.05, Mann-Whitney test). Median time to response with pramipexole was 4 weeks. Among the patients who completed the study, response rates tended to be higher among those taking pramipexole (seven of 10) than those taking placebo (one of six) (p=0.06, Fisher’s exact test). Remission, defined as a Hamilton depression scale score of 7 or less, occurred in two patients taking pramipexole and one patient taking placebo.
Mean CGI severity scores were lower at the end of the study for patients who took pramipexole (mean=2.7, SD=1.4) than for those who took placebo (mean=4.4, SD=1.3) (p=0.02, Mann-Whitney test). Improvement in CGI severity scores from baseline to study end was also significantly greater for patients who took pramipexole (mean=–2.4 points, SD=1.8) than placebo (mean=–0.30 points, SD=1.3) (p=0.01, Mann-Whitney test).
Lack of efficacy led to premature study discontinuation more often among the patients taking placebo (three of four patients) than among those taking pramipexole (one of two). No patients dropped out prematurely because of adverse events except for one patient taking pramipexole who developed mania with psychosis at week 6, despite concomitant divalproex (1000 mg/day, serum valproate=86 μg/ml upon termination). No patients taking placebo became manic.
Mean Young Mania Rating Scale scores at the end of the study did not differ significantly between the patients taking pramipexole (mean=4.4, SD=4.6) and those taking placebo (mean=2.0, SD=2.2) (p=0.12, Mann-Whitney test). Nausea tended to occur more often with pramipexole than placebo (seven [58%] of 12 versus two [20%] of 10, respectively) (p=0.10, Fisher’s exact test). Other adverse events common with pramipexole included sedation (N=3 [25%]) and headache (N=3), but proportions did not differ significantly from placebo.
Discussion
This pilot study preliminarily demonstrates antidepressant efficacy for the dopamine agonist pramipexole in treatment-resistant bipolar depression. Pramipexole was safe and effective when combined with lithium or anticonvulsants, consistent with the results of case reports and open trials
(7–
9).
Although few patients achieved full remission within 6 weeks, longer treatment durations might be necessary for optimal benefits. The vast majority of patients who openly took pramipexole after study completion showed marked responses that were sustained at least through 12-week follow-up. Our impression was that faster dose escalations (e.g., 0.25 mg every 2–3 days) might accelerate time to response unless limited by gastrointestinal or other adverse effects. Nausea, while frequent and usually persistent until protocol cessation, was generally mild and manageable by coadministration with food or over-the-counter remedies.
Limitations of the current study include the small number of subjects, inclusion of both bipolar I and bipolar II patients, random assignment of more women to pramipexole than to placebo, and continuation of varied prestudy medications. It is possible that pharmacodynamic synergies occurred by combining pramipexole with different agents, although all patients had been nonresponsive to previous treatments. In addition, heterogeneity of the group of patients studied might have affected outcome through some unidentified unmatched factor. The lower completion rate with placebo underscores patients’ symptom severity, although greater efficacy and adverse effects with pramipexole could also have inadvertently compromised the double-blind.
These preliminary findings suggest high tolerability and safety with pramipexole in bipolar depression. Larger-scale controlled trials are needed to affirm these initial observations.