The mechanism by which sleep deprivation exerts its antidepressant effect is still unknown. Presently, there is no commonly accepted hypothesis concerning the mechanism of action of sleep deprivation nor an explanation for the observation that subsequent sleep after sleep deprivation leads to relapse.
One of the most robust biological abnormalities in depression is an altered regulation of corticotropin (ACTH) and cortisol secretory activity in the majority of patients (for overview, see reference
4). It has been suggested that changes in the corticotropin-releasing factor (CRF) systems are a crucial aspect in the pathophysiology of major depression
(5) (for an overview, see reference
6). In addition, it has been suggested that antidepressants act through normalization of the hypothalamic-pituitary-adrenal (HPA) axis dysregulation
(7) (for a review, see reference
8). This hypothesis is confounded by the fact that many antidepressants have a direct stimulatory effect on the activity of the HPA axis independent of psychopathological state
(9–
12). Therefore, it is of interest whether therapeutic sleep deprivation—especially if successful—interferes with the activity of the HPA axis in depressed patients.
Up to now, the studies on the effect of sleep deprivation on HPA axis activity were mainly performed with healthy subjects. Whereas earlier studies showed little effect of sleep deprivation on the HPA axis
(13,
14), later investigations demonstrated a significant elevation of cortisol after sleep deprivation
(15). Vgontzas and co-workers
(16) reported a significant reduction of cortisol after sleep deprivation in healthy humans during the postdeprivation nighttime period. These authors proposed that a reduction of CRF and cortisol secretion might be the mechanism through which sleep deprivation relieves depression temporarily. However, in their study cortisol was not measured during sleep deprivation but in the postdeprivation night. Patients with major depression who improve after sleep deprivation usually have a relapse after the postdeprivation night. To our knowledge, up to now there have been no studies of depressed patients in which HPA activity was measured not only during a night of sleep deprivation or the day after but also during subsequent sleep.
It was therefore the primary objective of this study to determine precisely the impact of total sleep deprivation on cortisol secretion in patients with major depressive disorder during the night of sleep deprivation and the ensuing 24 hours in comparison to baseline conditions before sleep deprivation. The hypothesis was that the HPA axis is dampened by sleep deprivation in patients who respond to sleep deprivation but quickly returns to baseline levels during the recovery night.
Method
The study protocol was approved by the ethical committee of the University of Freiburg. Written informed consent was obtained from all subjects before study entry, after the procedure had been fully explained. We included 19 depressed inpatients, age 19 to 58 years, who were diagnosed as having major depression according to the Structured Clinical Interview for DSM-III-R
(17) and who scored at least 18 points on the Hamilton Depression Rating Scale
(18). The exclusion criteria were intake of any psychotropic drugs during and at least 1 week before the study, substance abuse, suicidality, history of endocrine disorders, pregnancy, postpartum depression, lactation, and any sleep disorder other than depression-related insomnia. The drug-free interval was longer than 4 weeks for all but one patient, who stopped doxepin intake 7 days before the study. Four patients dropped out, one because of problems with the blood withdrawal procedure and two because of exclusion criteria that had not been recognized at the time of inclusion. One patient withdrew her consent during the study, before the sleep deprivation night. Another patient felt worse on the morning after the sleep deprivation night, and her participation was terminated prematurely. However, her endocrine data were included in the analysis. The remaining patients (nine female and six male patients) had a mean age of 34.1 years (SD=13.4). The mean score on the 21-item Hamilton depression scale at study entry was 24.0 (SD=5.8), and the mean body mass index was 22.6 kg/m
2 (SD=3.7). Ten of the 15 patients reported diurnal variations of mood, all with improvement in the evening.
Each patient was given a physical and neurological examination, routine serum chemistry and hematology studies, an ECG and EEG, and a urine analysis to measure intake of illicit drugs or benzodiazepines.
Procedures
The study was carried out in the sleep laboratory of the Department of Psychiatry and Psychotherapy of the University of Freiburg. On nights 1 and 2, the patients were allowed to adapt to the laboratory conditions, and baseline polysomnographic sleep measures were obtained.
During nights 3, 4, and 5, blood was sampled at 30-minute intervals from 9:30 p.m. until 8.30 a.m. the next morning. The samples were collected through an indwelling catheter placed in a forearm vein 3 hours before the beginning of blood sampling on each of the 3 days. Between samplings the catheter was perfused by isotonic saline at a low speed (25 ml/hour). Between 11:00 p.m. and 7:00 a.m. the samples were taken from an adjoining room through polyethylene tubing. The total amount of blood collected per night was 110 ml.
From the evening of day 3 until the morning after the last night, the subjects stayed in the research ward. During the 40 hours, the experimental conditions were standardized as follows: standardized meals at 8:00 a.m., 12:30 p.m., and 6:00 p.m. on both days, no major physical activities, and a semisupine position in bed or a sitting position in a large armchair, which allowed reading or watching television. The subjects were strictly advised not to sleep and were therefore constantly observed by a team of three research fellows (including J.J.).
On days 4 and 5, saliva was collected at 30-minute intervals from 8:00 a.m. until 10:00 p.m. by using small cotton swabs that the subjects chewed for 30–60 seconds (for details of this method, see reference
19).
Serum and saliva cortisol levels were measured by radioimmunoassay using commercial kits with intra- and interassay coefficients of variations of less than 10%. All measurements were made in duplicate. The serum and saliva samples were stored at –80°C.
Sleep recordings were performed during all 4 nights from “lights out” (11:00 p.m.) to “lights on” (7:00 a.m.) according to standard criteria (the method of polysomnography in our laboratory is described in reference
20).
Depression Ratings
The 21-item Hamilton depression scale was administered at the beginning of the investigation
(18). Since the 21-item version is not suitable for repeated measurements in a sleep deprivation experiment, an abbreviated version with six items was used to measure depressive mood on the days before and after the sleep deprivation night as well as on the day after the recovery night at 9:00 a.m. and 6:00 p.m. The six-item Hamilton depression scale has a maximum score of 22 points and includes the following items: depressed mood, guilt feelings, decreased work ability and interest in usual activities, psychomotor retardation, anxiety, and physical symptoms
(21). Antidepressant response to sleep deprivation was defined as at least a 30% improvement in the average of the 9:00 a.m. to 6:00 p.m. scores on the six-item Hamilton depression scale on the day after sleep deprivation compared with the day before sleep deprivation.
Statistical Analysis
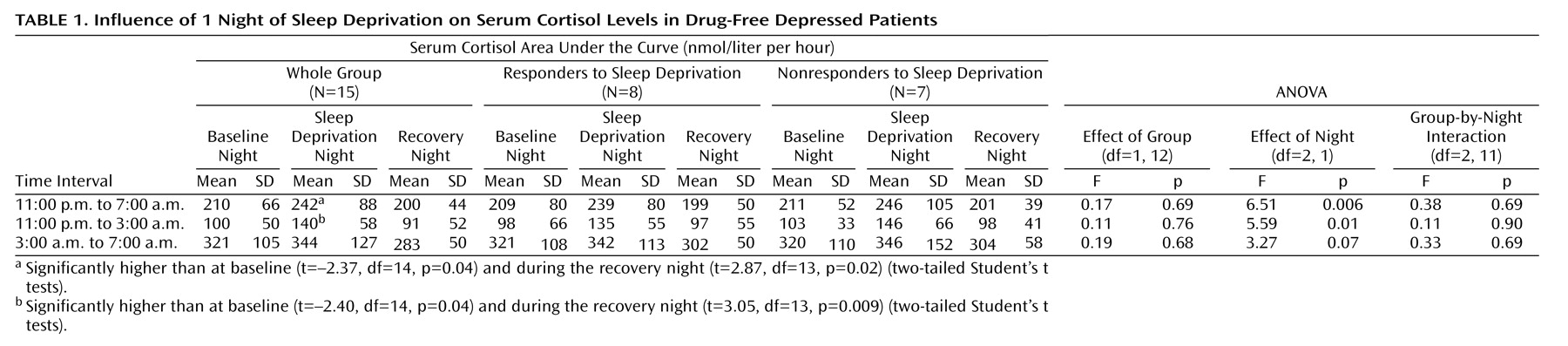
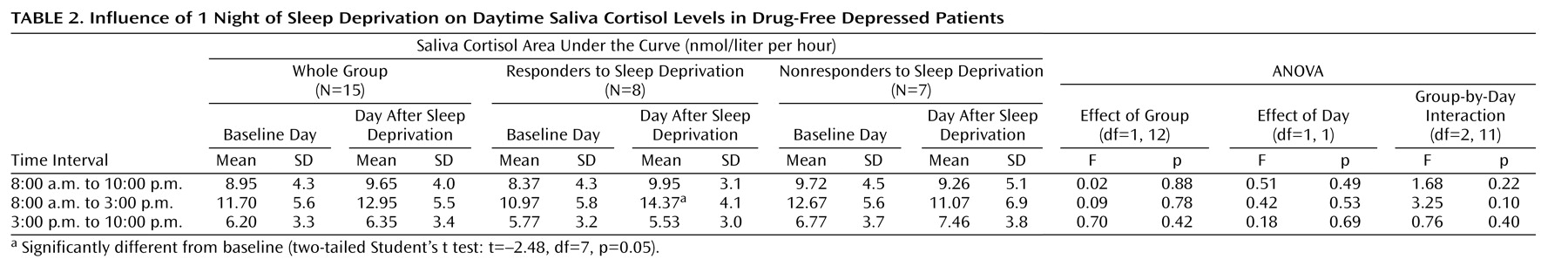
For descriptive purposes, means and standard deviations were calculated. To compare cortisol levels during and after sleep deprivation with baseline values, the area under the curve was calculated as the sum of all concentration values within the selected time interval (the first and the last value divided by 2) multiplied by the sampling interval (0.5 hours). Areas under the curve were calculated for the interval from 11:00 p.m. to 7:00 a.m., which corresponds to the registration time of polysomnography in nights 3 and 5, and for first and the second halves of the night, i.e., from 11:00 p.m. to 3:00 a.m. and from 3:00 a.m. to 7:00 a.m., respectively. Areas under the curve for saliva cortisol secretion during the daytime were calculated for the whole period between 8:00 a.m. and 10:00 p.m. as well as for the first half and the second half of the day, i.e., from 8:00 a.m. to 3:00 p.m. and from 3:00 p.m. to 10:00 p.m., respectively. Statistical analysis was performed by analysis of variance (ANOVA) for repeated measurements, and group (responders versus nonresponders to sleep deprivation) was used as a factor. In the case of statistically obvious differences (p<0.10), two-tailed Student’s t tests for dependent samples were calculated for comparisons of the 3 consecutive nights and of the days before and after sleep deprivation; p<0.05 was considered to be significant.
Discussion
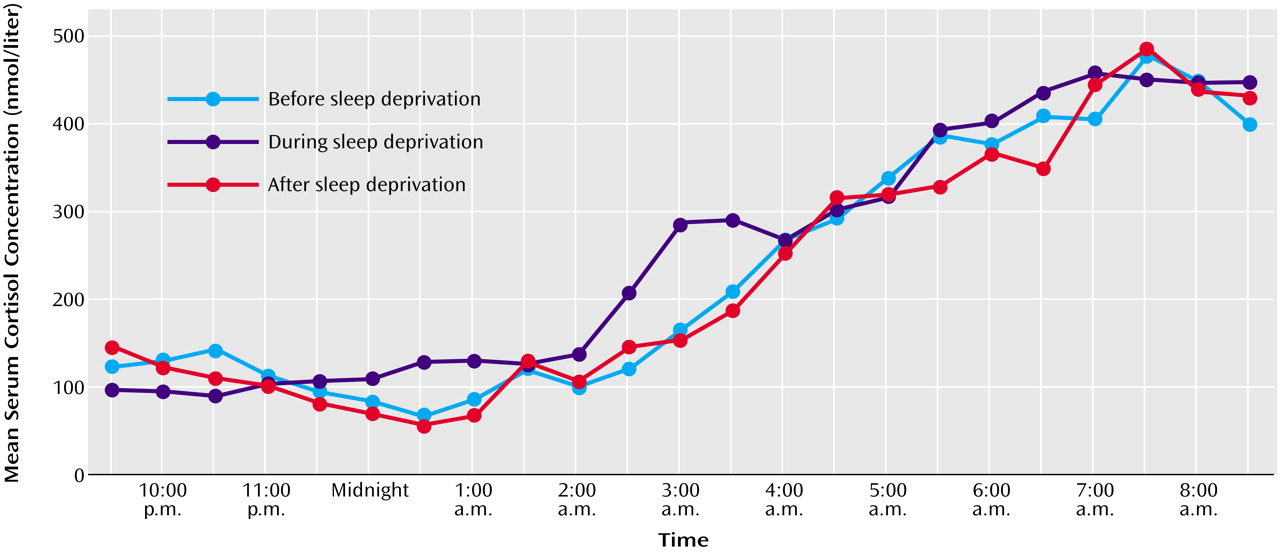
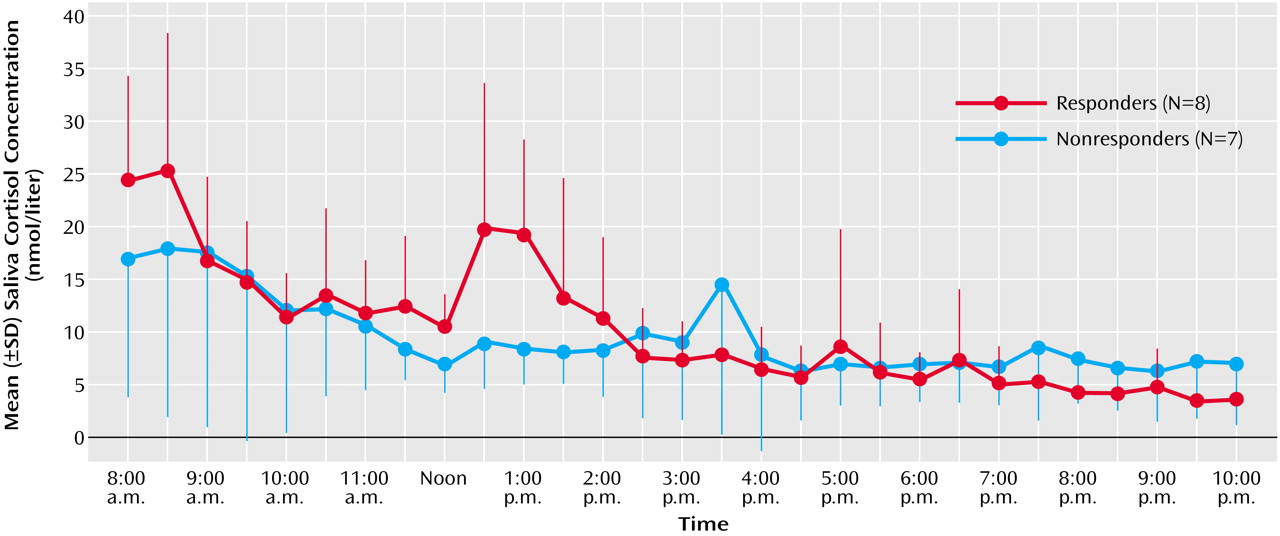
This study demonstrates that 1 night of therapeutic sleep deprivation slightly but significantly increases nocturnal cortisol secretion in patients with a major depressive episode. In the group as a whole, sleep deprivation did not enhance integrated cortisol levels during the daytime to levels higher than those on the day before. The responders to sleep deprivation, however, exhibited a significant rise of cortisol during the first half of the day, whereas nonresponders did not.
Regardless of the fact that our results were obtained in depressed patients, who might differ from healthy humans with respect to some functional aspects of the endocrine system, the stimulatory effect of sleep deprivation on the HPA axis in general is in line with earlier findings in healthy humans. These also demonstrated significant increases of cortisol secretion in response to total sleep deprivation for 1 night
(15,
22–24). The magnitude of cortisol elevation in the depressed patients was similar to that found in healthy subjects
(23,
24, and our own unpublished data). As in our responders to sleep deprivation, in healthy subjects the cortisol elevation persisted the next day
(23,
24), whereas in the study by Leproult and co-workers
(15) a significant increase of cortisol was observed only in the evening after sleep deprivation.
Our results are not consistent with earlier findings on the impact of sleep deprivation on the HPA axis in depressed patients. The results in many of these studies, however, are not comparable with those in the present investigation because of methodological factors. Some investigators took one blood sample after sleep deprivation
(25,
26). In some studies, patients were not free of medication
(26,
27). Parry and co-workers
(28) investigated the effects of sleep deprivation on cortisol in subjects with premenstrual dysphoric disorder and in normal comparison subjects and did not find quantitative effects on cortisol secretion. However, they performed only partial sleep deprivation, in the first or second half of the night. Goetze and Toelle
(29), who investigated depressed patients receiving medication, measured the 24-hour rhythms of free urinary cortisol over 5 days, including 1 night of sleep deprivation, and found enhanced cortisol levels in response to sleep deprivation. A similar finding was reported by Bouhuys and co-workers
(30), who obtained urine samples every 3 hours over a 24-hour period after 1 night’s sleep deprivation in unmedicated patients and found an increase of cortisol, which, however, was not related to clinical response. In a second study by Baumgartner et al.
(31), cortisol was measured during a night of sleep deprivation. An increase in cortisol was found in responders but not in nonresponders to sleep deprivation. A similar result, yet in a very small group of depressed patients, was also reported by Gerner et al.
(32). We believe that our study is the first to compare cortisol concentrations from frequent sampling in 3 consecutive nights before, during, and after sleep deprivation in unmedicated depressed patients. In terms of a general activation of HPA activity following sleep deprivation, our results agree with the findings of Goetze and Toelle
(29) and Bouhuys et al.
(30).
We did not find that cortisol secretion during recovery sleep was significantly lower than during the baseline night. Therefore, we could not confirm the hypothesis of Vgontzas et al.
(16), who reported a significant reduction of cortisol in healthy subjects during recovery sleep after sleep deprivation, in comparison to a baseline night before sleep deprivation, and who proposed that this decrease might be the mechanism through which sleep deprivation relieves depression temporarily.
Since sleep deprivation improves mood in depressed patients, it is noteworthy that the procedure activates the HPA axis over the short term, since remission of depressive symptoms is associated with a normalization of HPA axis dysfunction
(8,
33–35). The data from our study suggest that the short-term effects of antidepressant treatments on HPA activity may differ from, and may even be in the opposite direction from, their medium- or long-term effects. The same issue was also addressed by Kling et al.
(36), who measured HPA activity after electroconvulsive therapy (ECT) and found a significant increase of HPA activity in response to ECT, which also confirmed earlier findings by Aperia et al.
(37) and Rudorfer et al.
(38). One might assume that initial activation of HPA activity leads to a transient enhancement of negative feedback on the axis, leading to reduced CRF secretion. This assumption is supported by the findings of Posener et al.
(39), who demonstrated that, contrary to an earlier hypothesis, cortisol feedback in the HPA axis is not impaired in patients with major depression.
The trend observed in this and earlier studies that initial HPA activation is associated with therapeutic response to sleep deprivation is still preliminary and should be confirmed in large groups of subjects. At least there was definitely no trend in the opposite direction, i.e., no reduction of cortisol secretion in patients who showed an improvement of mood after sleep deprivation. It remains speculative whether HPA effects are related to the mechanism of the therapeutic action of sleep deprivation. The fact that a nocturnal elevation of cortisol was present in both nonresponders and responders to sleep deprivation does not support a relationship. However, the responders showed a more sustained elevation, persisting during the first half of the next day. Some other findings support the view that an increase of HPA activity might be relevant for the therapeutic effect. It has been demonstrated that hydrocortisone infusion but not placebo or CRF infusion has a short-term antidepressant effect, as does sleep deprivation
(40). Also, an earlier investigation by Goodwin et al.
(41) demonstrated short-term antidepressant effects of cortisol infusion in depressed patients, which was the opposite of what the authors expected. Another important issue is the functional coupling of cortisol and psychostimulant-like effects. Earlier studies indicated an increase of dopamine, noradrenaline, and serotonin after sleep deprivation, i.e., neurobiological effects similar to those following the intake of psychostimulants, such as amphetamines (see reference
42 for overview). The rewarding effects of psychostimulants, however, are dependent on the HPA axis
(43). A cortisol increase following sleep deprivation might therefore mediate psychostimulant-like actions of increased aminergic neurotransmitter release. This view agrees with that of DeBattista and co-workers
(40), who stated that the antidepressant effects of hydrocortisone might be mediated by an interaction with neurotransmitters, such as dopamine.
In conclusion, this study demonstrates that—much as in healthy humans—sleep deprivation significantly activates the HPA axis in unmedicated depressed patients.