Recently, an initial genetic screen of Level 1 outcomes in the STAR*D cohort was reported
(4) . The study tested 768 markers in 68 candidate genes for reproducible association with treatment outcome in the first two-thirds of the cohort and followed up significant findings in the total cohort. Reproducible association was detected between treatment response and a marker in the HTR2A gene, which encodes a serotonin receptor previously implicated in antidepressant mechanisms. In the present study, results from the full set of 1,816 genotyped individuals screened with the same set of markers were examined. Additionally, 634 psychiatrically healthy comparison subjects were gentoyped. These data allow us to address the question of whether case groups defined by treatment outcome differ from each other genetically and from comparison subjects.
Method
Patients, Cohorts, and Phenotypes
Detailed descriptions of the study population
(2), methods of DNA extraction, phenotypic definitions, clinical outcomes of Level 1, and our initial report from the first wave of genotyping
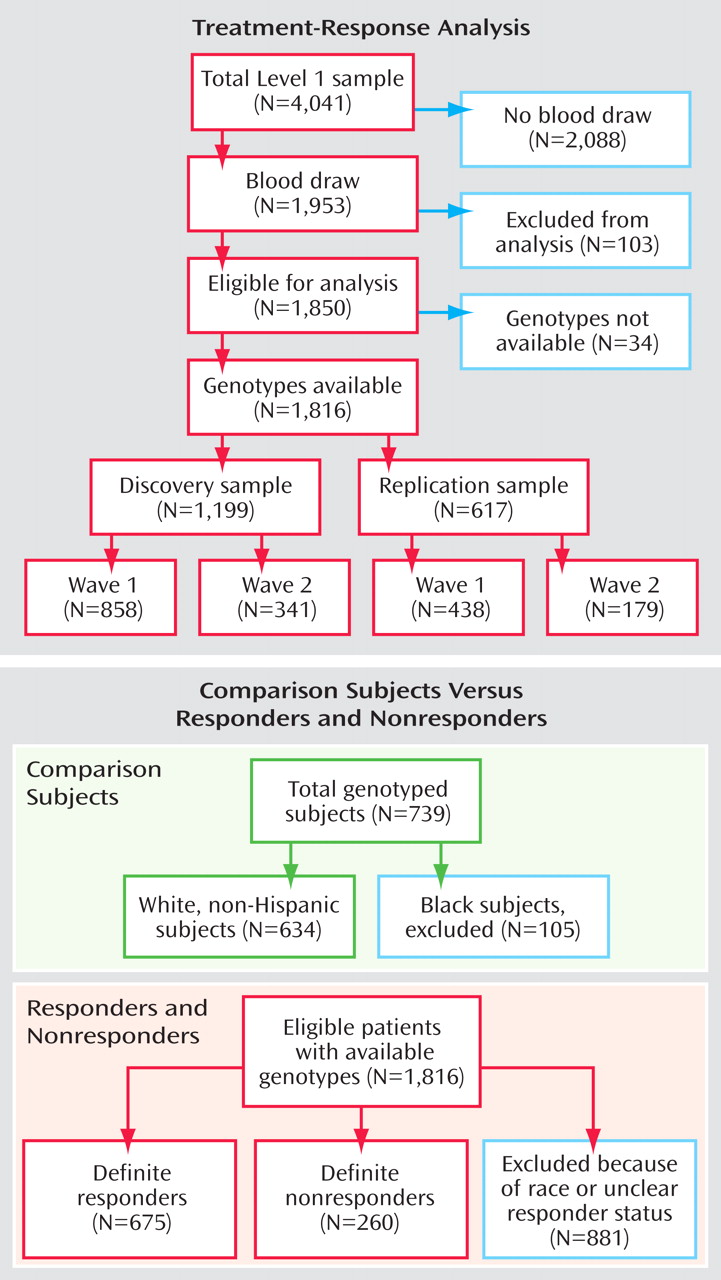
(4) are available elsewhere. In brief, DNA samples were available from 1,953 individuals affected by a major depressive episode and who sought outpatient treatment at one of 41 STAR*D clinical trial sites. A total of 103 DNA samples were excluded from analysis because of missing clinical data, noncompliance with the treatment protocol, or suspected sample mix-ups, leaving 1,850 samples for analysis. Of these, genotypes were available for 1,816 individuals, on which the results reported in the present study are based (a CONSORT diagram is shown in
Figure 1 ). All subjects gave written informed consent to participate in the clinical study and a separate written informed consent for collection and analysis of their blood samples. The study was supervised by the institutional review boards at all participating institutions and the Data Safety and Monitoring Board at the National Institute of Mental Health (NIMH). All definitions of primary outcomes were agreed upon before commencing genotype analyses.
Participants were enrolled without regard to race or ethnicity. Self-reported race and ethnicity data were available. For the purposes of this study, race was collapsed into “white,” “black,” or “other;” ethnicity was taken as reported (“Hispanic” or “Non-Hispanic”).
Severity of depression was assessed using the 16-item Quick Inventory of Depressive Symptomatology-Clinician Rated
(5,
6) . Individuals with a baseline score of 10 or more were eligible for these analyses. All patients received citalopram. Anxiolytics, sedative hypnotics, and other medications for concomitant general medical conditions were the only additional medications allowed.
To allow for treatment drug intolerance, we grouped participants based on reported side effects. In general, if participants left the study or refused to continue the same medication in a later stage and indicated side effects as the reason, they were considered “intolerant.” If they were willing to continue the medication in follow-up or in a later study level, they were considered “tolerant.” If neither of these situations applied, a designation of “probably tolerant” or “probably intolerant” was assigned based on the degree of side effect burden reported or, in the few persons for whom the data was unavailable, the length of time they continued receiving the medication. Further details on tolerability can be found in the initial genotyping report
(4) .
Primary outcomes of the treatment were “remission,” defined by an exit score of 5 or less, and “response,” defined by a reduction of at least 50% on the Quick Inventory of Depressive Symptomatology-Clinician Rated at the last visit receiving citalopram. To avoid misclassification, individuals with an exit score of 6 to 9 were excluded from both the “nonremitter” and “remitter” groups. Similarly, “nonresponders” were defined by a reduction of less than 40% from the baseline score. Individuals with at least a 40% but less than 50% reduction were excluded from all analyses presented in this study. Patients who completed less than 6 weeks of treatment were also excluded from analyses of these outcomes. Additionally, participants who did not respond to treatment but were classified as “intolerant” or “probably intolerant” were excluded. However, subjects who responded to treatment but who were intolerant (or probably intolerant) were included in the analyses. The rationale for this asymmetry was that intolerant patients may have dropped out of the study if they did not see any benefit of the treatment, while those with some improvement of depressive symptoms may have remained compliant despite any side effects. Relative reduction ([startscore-endscore]/startscore) of the Quick Inventory of Depressive Symptomatology-Clinician Rated score during the course of the trial was also analyzed as a quantitative trait.
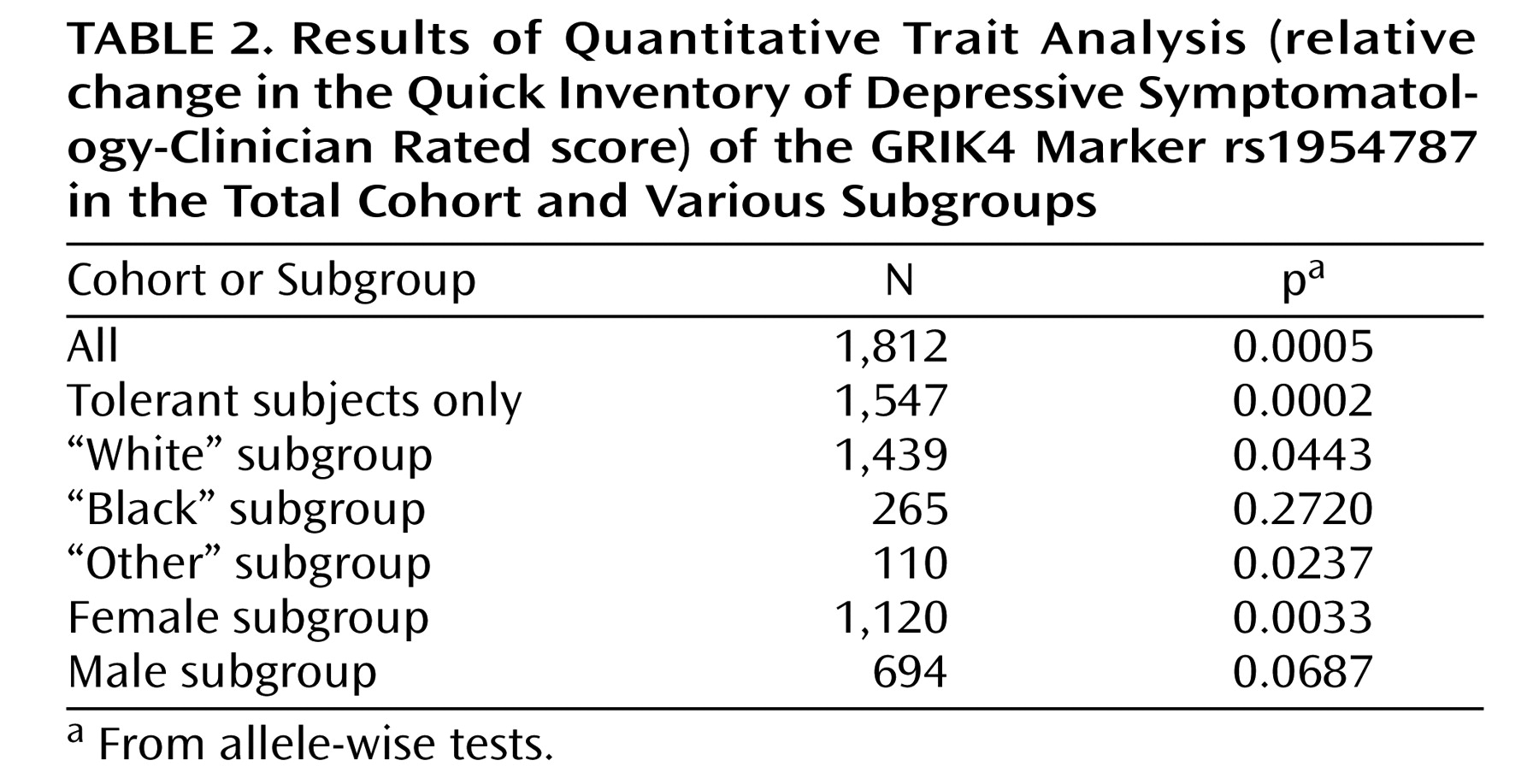
Exploratory analyses were conducted on alternative cohort subsets. These included tests of association in tolerant (or probably tolerant) subjects only, tests in men and women separately, and tests in cohorts from white or black participants only.
Comparisons
Comparison DNA samples (N=739) were obtained from the Rutgers DNA repository. These comparisons were collected via population sampling after participants provided written informed consent, under the auspices of the NIMH Genetics Initiative for Schizophrenia, and completed a basic psychiatric screening. Individuals who met DSM-IV criteria for major depression or reported a history of bipolar disorder or psychosis were excluded.
Within the comparison cohort, 105 individuals were self-reported as “black.” These individuals were excluded from the analyses in this study. All remaining individuals in the comparison study group identified themselves as “white, non-Hispanic.” Thus, all comparisons between comparison subjects and the STAR*D patients are confined to patients who self-reported as “white, non-Hispanic” (
Figure 1 ).
Candidate Genes and Marker Selection
Altogether, 768 markers covering 68 candidate genes were studied. The complete list of genes and the criteria for marker selection have been published previously
(4) . Genes were chosen based on their role in major neurotransmitter systems or other pathways thought to be involved in antidepressant outcome or mood disorders and were prioritized if functional variants were known. The gene list was completed by adding known members of targeted gene families (e.g., additional isoforms of serotonin or glutamate receptor subunits). Each gene was covered with all available markers from the HapMap Phase I
(7), with a minor allele frequency of 7.5% or greater. To avoid redundancy, markers with a pairwise r
2 correlation of ≥0.8 with any other selected marker
(8) were not genotyped. Six nonsynonymous single nucleotide polymorphisms and four single nucleotide polymorphisms previously implicated in response to antidepressant treatment were added
(9,
10), resulting in 768 markers.
Genotyping and Quality Control
All 1,953 patient DNA samples and 739 comparison DNA samples were shipped to Illumina Inc. (La Jolla, Calif.) and genotyped on the highly accurate Bead-Array platform
(11) . The genotyping success rate was 99.90%, and 99.73% of samples were successfully genotyped. Overall, 35,052 duplicates were analyzed by Illumina, and 878 samples were typed for four markers, both at Illumina and in the NIMH laboratory. No discrepancies were detected.
Statistical Analysis
We followed the analysis plan previously described
(4), but included the full set of 1,816 eligible individuals who were genotyped by Illumina. In the present study, identification of the previously identified HTR2A gene association was not considered to constitute a replication or confirmation of the earlier finding, since the same analysis plan was followed with the same markers and DNA samples. Instead, the present results are an extension of the previous study, completed with a maximum available power in the total cohort of 1,816 individuals. The primary outcomes were response and remission, as previously defined in this article. As in the previous study
(4), the cohort was divided into a “discovery” group, comprising two-thirds of the cohort, and a “replication” group, comprising the remaining one-third. The two study groups were matched for sex and ethnicity (“white,” “black,” or “other”). Each categorical outcome was tabulated against each marker and was coded in the following three ways: 1) presence versus absence of allele 1, 2) presence versus absence of allele 2, and 3) the three-valued genotype. For each analysis, Fisher’s two-sided exact (for allele-wise tests only), Pearson’s chi-square, and the likelihood ratio chi-square tests were considered. These analyses were implemented in the SAS FREQ Procedure (SAS Institute Inc., Cary, N.C.). Fisher’s exact test provided the most conservative results. Therefore, the results of the treatment-response association reported in the present study are based only on the Fisher’s exact test. Based on power analyses
(4), we considered a p≤0.01 in the discovery study group and a p≤0.05 in the replication study group to be a significant association, provided that the direction of the association was consistent between the two groups. For the quantitative trait, relative reduction in Quick Inventory of Depressive Symptomatology-Clinician Rated, analyses of variance (ANOVAs) were performed as implemented in the SAS ANOVA procedure (SAS Institute Inc., Cary, N.C.).
For exploratory analyses (in subgroups based on tolerability, sex, or race), findings in the present study cleared the predefined levels of significance in at least one of the three tests (Fisher’s two-sided exact [for allele-wise tests only], Pearson’s chi-square, and the likelihood ratio chi-square tests). Case-control association tests between responders and comparison subjects and between nonresponders and comparisons subjects were conducted using the COCAPHASE program from the UNPHASED suite of software, which generates p values based on a likelihood-ratio test
(12) .
Receiver operating characteristic analyses were carried out using logistic regression implemented in SAS (SAS Institute Inc., Cary, N.C.) to examine the effects of markers on the treatment-response phenotype. All p values reported in the present study are two-tailed. Pairwise linkage disequilibrium (expressed as r
2 =value) was calculated using HAPLOVIEW (version 3.2)
(13) .
Population Structure
STRUCTURE
(14) was used to test for the presence of cryptic population structure within the “white” subset. The program was run for 20,000 burn-in steps and 20,000 replications. Compared with our earlier study, the number of markers was increased to 301 single nucleotide polymorphisms, with an r
2 ≤0.3 between the markers. To account for any structure, we used the posterior probabilities of membership in each putative cluster as covariates in the association analysis
(15), which was performed with UNPHASED 3.06 (http://www.mrc-bsu.cam.ac.uk/personal/frank/software/unphased/).
Discussion
The present study is an extension of our previous genetic analyses of the STAR*D cohort
(4) . With the full set of genotypes now available from all 1,816 eligible individuals, we could conduct a more powerful analysis. Compared with the earlier study, the discovery group was increased by 341 subjects, and the replication group was increased by 179 subjects (
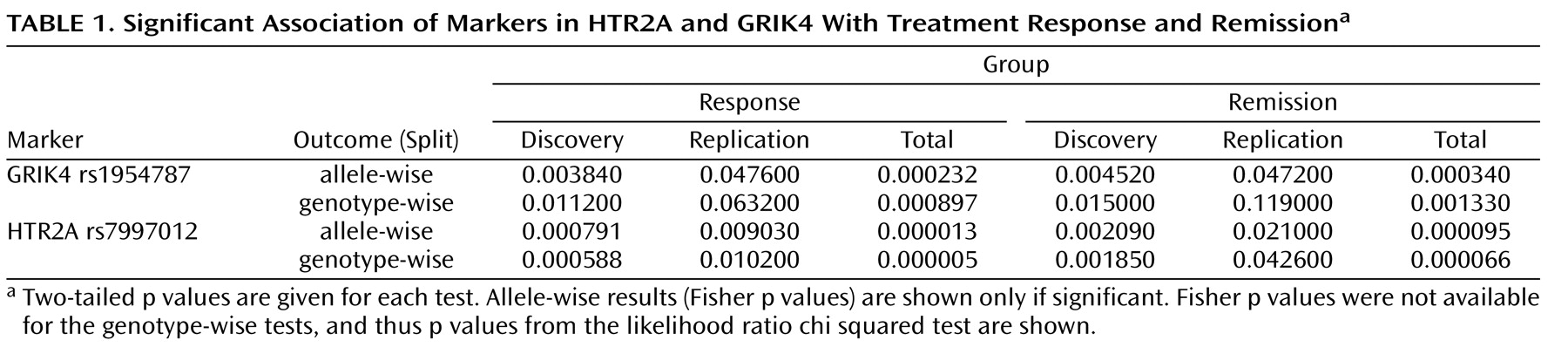
Figure 1 ). The present analyses identified an additional marker (rs1954787) associated with treatment response and remission. This marker is located in the 3′ end of the first intron of the GRIK4 gene. The marker does not alter the protein sequence but may have functional relevance in the regulation of gene expression. A search of the TRANSFAC database of transcription factor binding sites (http://www.gene-regulation.com/pub/databases.html#transfac) revealed a putative CCAAT enhancer element at the site of the polymorphism that is disrupted in the minor allele. Additionally, the marker rs1954787 maps to a bacterial artificial chromosome clone to which a chromosomal breakpoint has been mapped in a patient with schizophrenia
(17) . This suggests that the single nucleotide polymorphisms may be in linkage disequilibrium with a consensus breakpoint sequence. The marker may be in linkage disequilibrium with a functional variant that remains to be identified.
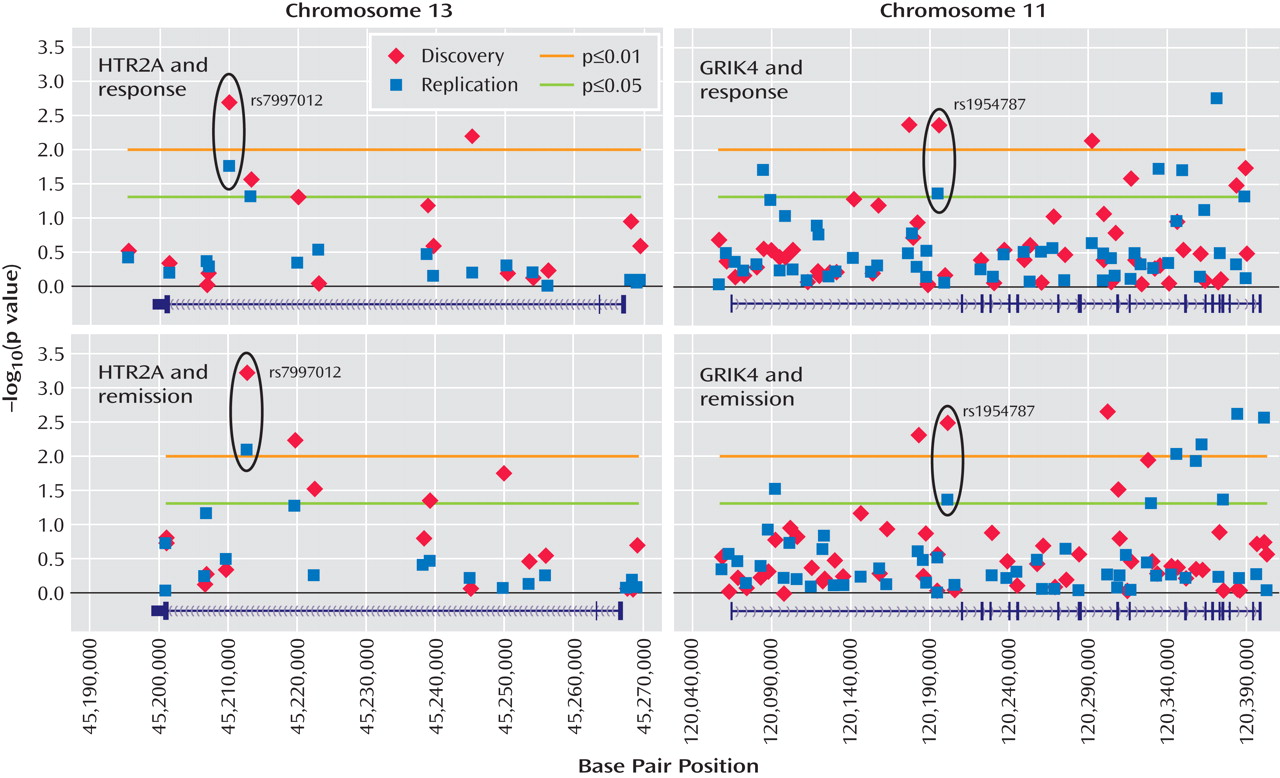
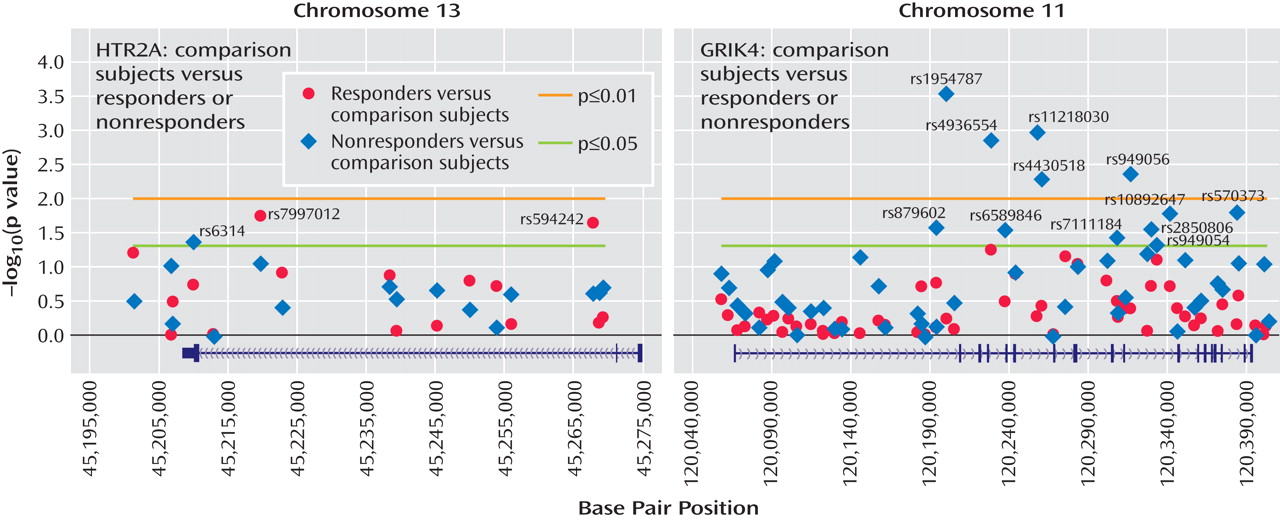
The healthy comparison group (N=634) allowed us to address, for the first time, questions concerning the nature of the observed associations with treatment response. For the HTR2A gene, the previously reported marker, rs7997012, primarily distinguished treatment responders from comparison subjects (
Figure 3 ). Two other significant associations were found in this gene: one marker differed from comparison subjects in the responders only, and one marker differed from comparison subjects in the nonresponders only (
Figure 3 ).
In the GRIK4 gene, we found that only nonresponders differed significantly from healthy comparison subjects in allele frequencies (
Figure 3 ). Significant differences were detected at 12 markers that were not strongly associated with each other. This suggests that several independent associations were detected and that common variation in the GRIK4 gene is associated with treatment-resistant depression. It is possible that comorbid conditions may contribute to this association. An initial search has identified anxious depression as one comorbid condition that is also associated with the GRIK4 gene (unpublished data by Laje et al. available from the authors). On the basis of a multiple regression analysis, however, it appears that both anxiety and treatment response showed significant independent associations with the GRIK4 genotype.
In the initial analysis of the STAR*D genetic cohort, intolerant and probably intolerant patients were excluded from the nonresponder group but not from the responder group
(4) . This may have introduced an asymmetry that could increase association findings driven by association with tolerance rather than treatment response. We therefore conducted a secondary analysis in which only tolerant and probably tolerant patients were included. Association of the rs7997012 marker remained significant, as previously reported. Association of the newly identified GRIK4 marker rs1954787 also remained significant. Thus, we conclude that these markers are related to treatment response rather than to intolerance.
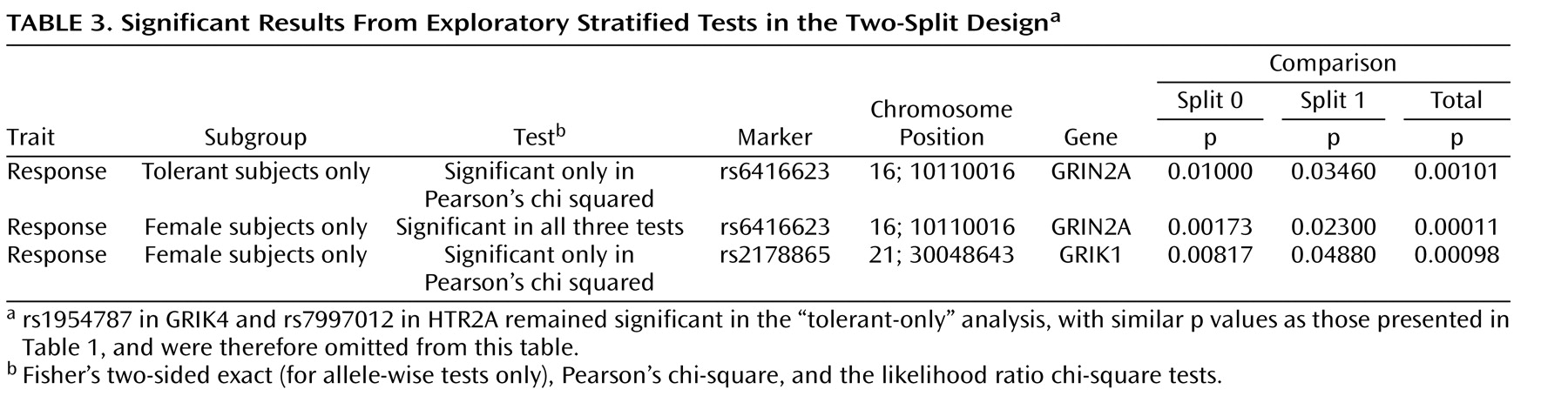
In the present study, we also report exploratory analyses that suggest other genes that may be involved in treatment outcome, but these findings do not hold up to stringent correction for multiple testing, and thus should be considered preliminary. Association was observed with the rs6416623 marker in GRIN2A, a gene coding for an
N -methyl-
d -aspartate-type glutamate receptor. Association was also detected between treatment response and markers in GRIN2A and GRIK1, but only in female patients. These findings are consistent with the hypothesis that major depression may be related to some distinct genes in male versus female patients
(18) . Treatment response may follow a similar division between sexes.
The most common hypothesis for major depressive disorder is still the biogenic amine hypothesis, which proposes that decreased synaptic availability of monoamine transmitters results in a condition that is reversed with antidepressant treatment. Excitatory amino acid neurotransmission, of which glutamate is the major example, may also play a role in major depressive disorder. Glutamate levels are increased in patients with major depressive disorder by proton magnetic resonance spectroscopy
(19), suggesting enhanced glutamatergic transmission. Ionotropic glutamate receptors, such as the GRIK4-encoded kainic acid-type subunit, are responsible for fast-acting neurotransmission. Ionotropic glutamate receptors are present presynaptically and postsynaptically, where they modulate neurotransmitter release or excitatory neurotransmission. A role for ionotropic glutamate receptors in antidepressant action has been supported by studies in rodents in which chronic treatment with antidepressants, including citalopram, resulted in a region-specific reduction in binding activity of
N -methyl-
d -aspartate receptors
(20,
21) and
N -methyl-
d -aspartate receptor subunits
(22) . Chronic treatment of rats with SSRIs under a regimen similar to that used in humans has been reported to attenuate glutamatergic transmission in the cerebral cortex
(23) . Finally, in a recent randomized placebo-controlled study, intravenous injection of ketamine, an
N -methyl-
d -aspartate receptor antagonist, led to rapid improvement of depressive symptoms in subjects with major depression
(24) . In the hippocampus, another brain region thought to be affected in patients with major depressive disorder, presynaptic KA receptor subunits (GluR5-7, KA1, and KA2 [also referred to as GRIK1-5]) regulate glutamate release. Activation of presynaptic KA receptors results in long-lasting inhibition of neurotransmitter release
(25) . This response may offset enhanced glutamatergic neurotransmission, constituting a feedback mechanism. Our finding that variation at a marker in the GRIK4 gene is associated with nonresponse to citalopram treatment suggests a role for KA receptor-dependent negative modulation.
To our knowledge, this is the first report of association between the GRIK4 gene and outcome of antidepressant treatment. A recent report indicated a possible association of GRIK4 haplotypes with schizophrenia and bipolar affective disorder
(17), spanning the same genomic region as the 12 markers in the present study. This suggests functional variation in this part of the GRIK4 gene, which may be of relevance beyond treatment response in major depression. Interestingly, decreased expression of the highly related GRIK1 and/or GRIK5 genes has been observed in postmortem brains of patients with major depression
(26) and bipolar disorder
(26,
27) .
In the present study, the relatively modest effect size of the GRIK4 association probably precludes any immediate clinical relevance. The associated “C-allele” reduces the absolute risk of nonresponse by 11% in homozygote carriers, but the c-statistic of 0.58 observed in combination with the previously reported response-associated HTR2A marker is modest. In the present study, only the two markers that met replication criteria were included in the interaction analysis. It is likely that many more markers will have to be included before more meaningful clinical findings emerge.
The demonstration that a glutamate-receptor encoding gene mediates antidepressant treatment response may provide novel clues to the mechanism of action of SSRIs and insight into critical downstream signaling cascades.