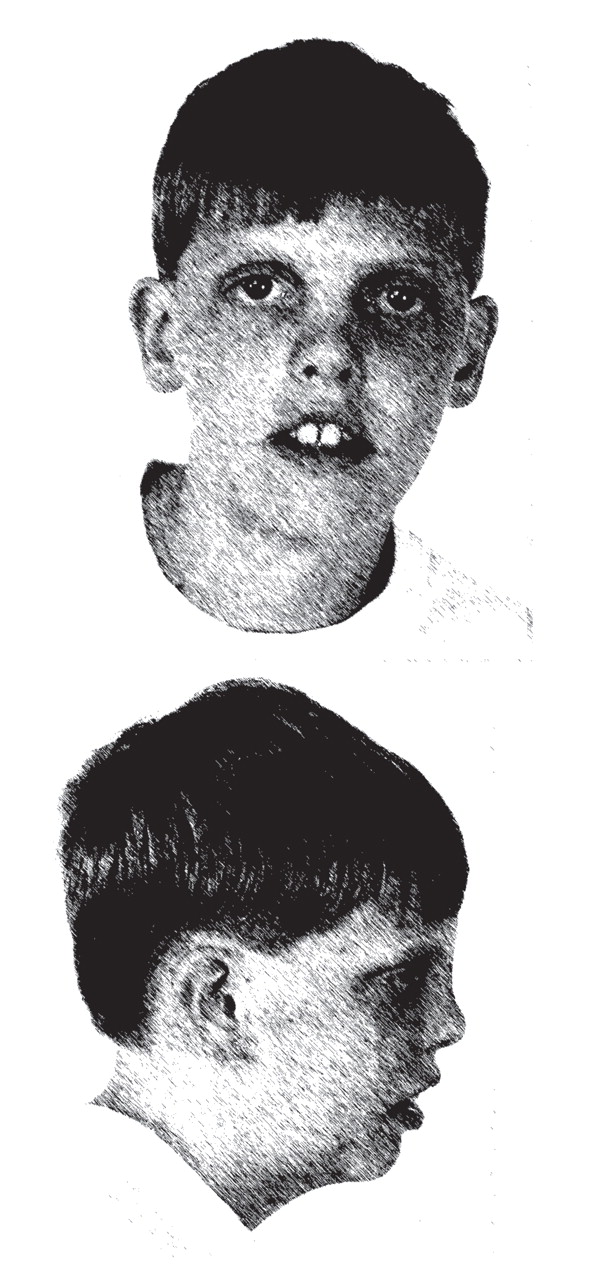
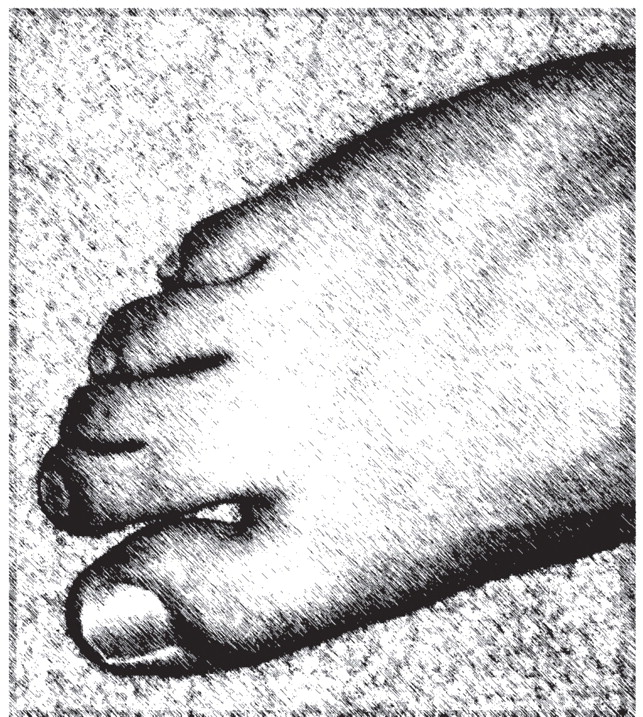
The authors discuss the case of a 9-year-old boy who came to the clinic with autism spectrum disorder, intellectual disability, impulsivity, and overactivity. He had bilateral ptosis, upturned nares, and two-to-three toe syndactyly. An inborn error of cholesterol synthesis, Smith-Lemli-Opitz syndrome, was identified. The patient was treated with dietary cholesterol supplementation. A report is presented to highlight the importance of suspecting this genetic disorder in the presence of autism spectrum disorder and dysmorphic facial features, minor anomalies, or specific behavioral phenotype features. The clinical features of Smith-Lemli-Opitz syndrome are reviewed, along with recommendations for diagnostic testing for the syndrome. The authors discuss how impairment in cholesterol synthetic pathways may result in abnormalities characteristic of autism spectrum disorder.
Autism spectrum disorder, a complex and enigmatic disturbance of brain function, is behaviorally defined by core areas of specific abnormalities in reciprocal social interaction, communication, and restricted or repetitive interests and behaviors
(1) . Autism spectrum disorder appears as a spectrum of conditions, including autism, pervasive developmental disorder not otherwise specified, Asperger’s disorder, childhood disintegrative disorder, and Rett’s syndrome
(1) . With an incidence of 1 in 150
(2), autism spectrum disorder becomes manifest in early childhood and usually persists throughout life. In the majority of cases, no specific underlying cause is identifiable at this time. However, a number of factors are being investigated, including infectious, metabolic, genetic, and environmental factors, with specific causes being documented in generally less than 10% of cases
(1) . Recently, a high prevalence (19%) of below normal levels of cholesterol was found in 100 autism spectrum disorder Autism Genetic Resource Exchange study participants who had a sibling with autism spectrum disorder
(3) . In addition to the association of autism spectrum disorder with specific heritable disorders (e.g., fragile X syndrome, phenylketonuria, chromosome 15q11-q13 duplication, and tuberous sclerosis), evidence for a genetic contribution includes increased recurrence risk in siblings; increased concordance in monozygotic compared to dizygotic twins; and occurrence of cognitive, language, and behavioral disturbances in close relatives
(1) .
Smith-Lemli-Opitz syndrome (MIM 270400)
(4) is an autosomal recessive disorder due to an inborn error of cholesterol synthesis caused by mutations of the 7-dehydrocholesterol reductase gene (
DHCR7 ) located on chromosome 11q12-13
(4) . Lack of normal
DHCR7 activity impairs the conversion of 7-dehydrocholesterol to cholesterol, causing an increased level of 7-dehydrocholesterol in blood and tissues and, in most patients, decreased blood and tissue cholesterol levels. Smith-Lemli-Opitz syndrome has an estimated incidence among individuals of European ancestry of 1 in 20,000 to 1 in 60,000 births and a carrier frequency of at least 1%
(4) . Smith-Lemli-Opitz syndrome is characterized by a broad spectrum of phenotypic abnormalities, including developmental delay, characteristic facial anomalies (microcephaly, bitemporal narrowing, ptosis, upturned nares, and a small chin), and abnormal webbing between the second and third toes (two-to-three toe syndactyly). Severely affected infants often die before birth or during the perinatal period
(4) . In mild cases of Smith-Lemli-Opitz syndrome, the physical phenotype may not be readily discernible, and toe syndactyly may not be present.
Autism spectrum disorder is among the most severe behavioral problems associated with Smith-Lemli-Opitz syndrome. Approximately 50% of individuals with Smith-Lemli-Opitz syndrome and a nonverbal mental age of 18 months or greater met the criteria for autism
(5) as diagnosed by both the DSM-IV and the Autism Diagnostic Interview—Revised algorithm questions
(6) . A recent study reported that approximately three-fourths of the children with Smith-Lemli-Opitz syndrome had met criteria for some variant of autism spectrum disorder, suggesting a consistent relationship with autism spectrum disorder and a single-gene disorder
(7) . Other behavioral phenotypic characteristics include cognitive delay, language impairment, self-injury with biting of the wrist or the thenar eminence, severe sensory hyperreactivity, irritability, sleep-cycle disturbance, and opisthokinesis (a highly characteristic upper body movement in which the individual performs an arched backward diving motion)
(5) . A clinical diagnosis of Smith-Lemli-Opitz syndrome can be confirmed by biochemical testing. An elevated plasma 7-dehydrocholesterol level relative to the cholesterol level establishes the diagnosis. Smith-Lemli-Opitz syndrome is not only identifiable, but it is also partially treatable by cholesterol supplementation. Thus, it is important to know the variations in the presentation of Smith-Lemli-Opitz syndrome so that individuals with autism spectrum disorder who have Smith-Lemli-Opitz syndrome can be identified and begin treatment.
We report on a boy with autism spectrum disorder and dysmorphic features who was diagnosed with Smith-Lemli-Opitz syndrome. A genetic cause of his autism spectrum disorder was not previously suspected. This case underscores the importance of recognizing that Smith-Lemli-Opitz syndrome is a distinctive and diagnostically important behavioral disorder that can be associated with autism spectrum disorder. In the discussion, we present our hypothesis that impairment in the cholesterol biosynthesis pathway can result in abnormalities that are characteristic of autism spectrum disorder. Furthermore, we review the clinical features of Smith-Lemli-Opitz syndrome and suggest when diagnostic testing for Smith-Lemli-Opitz syndrome should be performed.
Case Presentation
Family, Pregnancy, and Past Medical History
History of Present Illness
Biochemical and Molecular Testing
Autism Spectrum Disorder Assessment
Follow-Up Assessments After 1 and 2 Years
Discussion
This case illustrates many of the clinical features typically seen in individuals with autism spectrum disorder. Consistent with the description of autism spectrum disorder, in the context of Smith-Lemli-Opitz syndrome, C exhibited impairments in social interaction and communication, and he had repetitive behaviors. Although C’s diagnostic assignment could be considered relatively straightforward and characteristic of autism spectrum disorder, the recognition of a genetic cause of autism spectrum disorder was far more complex. C’s facial appearance and his two-to-three toe syndactyly were typical of Smith-Lemli-Opitz syndrome. Children with abnormal findings on physical examinations are 10 times more likely to have an underlying genetic condition than children with a normal physical examination
(1) . The physical and behavioral features of Smith-Lemli-Opitz syndrome are summarized in
Table 1 . Often individuals with a mild Smith-Lemli-Opitz syndrome phenotype are diagnosed because there is a relative diagnosed with Smith-Lemli-Opitz syndrome who has more prominent physical manifestations. C did not have a family member previously diagnosed with Smith-Lemli-Opitz syndrome. Because of the autosomal recessive nature of Smith-Lemli-Opitz syndrome, counseling and genetic testing was offered to the family. C’s brother, who did not have facial or other dysmorphology, tested negative for Smith-Lemli-Opitz syndrome by sterol analysis.
Failure to thrive and poor weight gain despite adequate caloric intake are common features of Smith-Lemli-Opitz syndrome, which were seen in C. Hypotonia, seen in C during his first years of life, is commonly reported in Smith-Lemli-Opitz syndrome and typically improves later in childhood. Infections, particularly otitis media, seem to occur more often in children with Smith-Lemli-Opitz syndrome (as was seen in C, resulting in mild conductive hearing loss). Behaviorally, C had sleep problems that are found in almost 70% of individuals with Smith-Lemli-Opitz syndrome
(5) . For most individuals, sleep-cycle problems persist to adulthood. C’s verbal abilities and lack of aggression were considered his strengths. Common behavioral problems that are classified under autism spectrum disorder include hand flapping, abnormal obsessions, rigidity and insistence on consistent routines, and poor eye contact. C met the diagnostic criteria for autism based on DSM-IV, the Autism Diagnostic Interview—Revised, and the Autism Diagnostic Observation Schedule—Generic. The Autism Diagnostic Interview—Revised requires a nonverbal mental age of 18 months or higher. Based on C’s previous IQ tests, C’s nonverbal mental age was above the required 18 months. Seizures, reported in C, occur in a third of individuals with autism
(1) . We acknowledge that the assessment instruments used did not assess the full range of C’s abilities.
The recognition of the biochemical cause of Smith-Lemli-Opitz syndrome not only significantly improved the diagnostic accuracy by measuring plasma levels of 7-dehydrocholesterol but also offered a potential treatment strategy. The current practice for treatment is to begin dietary cholesterol supplementation as soon as the condition is diagnosed. There are reports that cholesterol supplementation may improve sterol levels, growth, and neurodevelopmental status
(13) . In the case described here, the child responded very positively to cholesterol treatment, and his behaviors improved. His improvements may also be attributed to the speech and language therapy or to the educational services received. Currently, there is a new promising research treatment underway for Smith-Lemli-Opitz syndrome. Simvastatin treatment, which usually lowers cholesterol in a typical population, may stimulate residual
DHCR7 activity and, therefore, increase cholesterol production. One important feature of simvastatin is its ability to cross the blood-brain barrier as opposed to cholesterol, which does not.
Professional organizations are increasingly moving toward recommendations for screening for biochemical and genetic disorders that are associated with autism spectrum disorder, as described in the American Academy of Neurology and the Child Neurology Society’s
Practice Parameter: Screening and Diagnosis of Autism (14) . If a genetic disorder is suspected, the recommended diagnostic tests to evaluate a child for recognized causes of autism spectrum disorder (in the presence of mental retardation) include a lead level, a high-resolution chromosome study, and DNA analysis for fragile X syndrome. Based upon additional symptoms and clinical findings, tests to be considered include the following: an EEG for seizure activity evaluation, quantitative urinary organic and plasma amino acids, fluorescence in situ hybridization for Angelman’s syndrome, chromosome 15q11-q13 duplication and velocardiofacial syndrome, and analyses of serum carbohydrate-deficient transferrin to rule out congenital disorders of glycosylation
(1,
15) . In addition, gene sequencing of phosphatase and tensin homologue, deleted on chromosome 10 (
PTEN ), for patients with macrocephaly, autism, and developmental delay, as well as methyl CpG binding protein 2 (
MECP2 ) in girls suspected of Rett’s syndrome, is suggested
(16) . Because of a low yield of a specific genetic diagnosis (below 10%), we do not recommend routine testing for genetic disorders unless there is specific evidence to suspect a disorder.
Cholesterol-Related Mechanisms in Autism Spectrum Disorder
The autism rate in Smith-Lemli-Opitz syndrome (1:2) is much higher than in the general population (1:150), and it is among the highest when compared to other single-gene disorders, indicating that a genetic defect in Smith-Lemli-Opitz syndrome could be responsible for the autism spectrum disorder phenotype seen in Smith-Lemli-Opitz syndrome. Since the report that almost half of the individuals with Smith-Lemli-Opitz syndrome have autism
(5), there has been an increasing recognition that this single-gene disorder of abnormal cholesterol synthesis may be a model for understanding genetic causes of autism and the role of cholesterol in autism spectrum disorder
(7) . Furthermore, the detection of a high prevalence of abnormally low cholesterol blood levels in an autism spectrum disorder subject population is a finding of potential clinical significance regarding the possible role of non-Smith-Lemli-Opitz syndrome cholesterol disorders in the etiology of autism spectrum disorder
(3) . The sterol abnormality may affect the brain in different ways involving multiple genes in each individual, but there may be a common neurodevelopmental pathway that causes behaviors seen in autism spectrum disorder. In some forms of autism spectrum disorder, the core symptoms of autism spectrum disorder may be due to the interplay of components that are sterol dependent. The high prevalence of autism spectrum disorder in Smith-Lemli-Opitz syndrome reported by two independent studies
(5,
7) using slightly different methodologies indicates that these conditions might share etiological mechanisms. Listed below are our observations related to sterol function in Smith-Lemli-Opitz syndrome and hypotheses as to potential sterol mechanisms that might be involved in autism spectrum disorder.
CNS Development
Cholesterol is essential for embryonic and fetal development. Structural CNS abnormalities, similar to those observed in individuals with autism spectrum disorder, are present in individuals with Smith-Lemli-Opitz syndrome because of the malfunction of the hedgehog patterning protein that results from inadequate availability of cholesterol
(4) .
Myelination
An adequate cholesterol level is essential for myelin membrane growth. In mice, it was shown that cholesterol is an indispensable component of myelin membranes and that cholesterol availability in oligodendrocytes is a rate-limiting factor for brain maturation
(17) .
Serotonin Receptor and Transporter
Cholesterol functions as an intracellular modulator of the ligand-binding activity and G-protein coupling of the serotonin1A (5-HT
1A ) receptor
(18), and cholesterol is a component of the lipid rafts of the serotonin transporter
(19) . Disruption of lipid rafts by cholesterol-interfering agents produces a 50% decrease in the transport rate of the GABA transporter, 5-HT transporter, and glutamate transporters
(20) . It was also shown that suboptimal 5-HT transporter and GABA transporter activity occur when cholesterol is replaced with sterols, indicating specific cholesterol-transporter interactions
(20) . In addition, abnormal serotonergic neuron development has been demonstrated in a mouse model of Smith-Lemli-Opitz syndrome
(21) .
Steroid Hormones
Cholesterol is a precursor for neuroactive steroid production, and the alteration in steroid function in Smith-Lemli-Opitz syndrome may have a role in the clinical presentation of autism spectrum disorder. Neuroactive steroids exhibit a wide range of modulatory effects on neurotransmitter receptor activity and neurodevelopmental and neuroprotective effects and, if deficient, may be associated with mood and anxiety disorders
(22) . There is evidence for decreased neuroactive steroid levels, dehydroepiandrosterone, and dehydroepiandrosterone-sulfate in adult patients with autistic disorder
(22) .
Oxytocin Receptor
Cholesterol has a modulatory role on the function of the oxytocin receptor
(23), and oxytocin has been found to be involved in social function. It could be that low cholesterol perturbs the oxytocin receptor and thereby contributes to problems in social functioning in individuals with Smith-Lemli-Opitz syndrome as well as those with autism spectrum disorder from other etiologies.
Recommendations
Because specific testing and partial treatment with cholesterol supplementation for Smith-Lemli-Opitz syndrome are available, the threshold should be low for obtaining a sterol analysis for Smith-Lemli-Opitz syndrome in an individual with characteristic phenotypic features and autism spectrum disorder. The association of autism spectrum disorder with physical or behavioral manifestations that warrant testing for Smith-Lemli-Opitz syndrome include the following: two-to-three toe syndactyly, ptosis, soft cleft palate/bifid uvula, failure to thrive or feeding difficulties, growth retardation, postnatal onset of microcephaly, hand or foot malformation, abnormal genitalia, hypotonia, severe sleep disturbance, self-injury with biting of the wrist or the thenar eminence, opisthokinesis, or a family history of developmental disabilities with physical dysmorphology.
Conclusion
In summary, we presented a 9-year-old boy with autism spectrum disorder and intellectual disability who was later diagnosed with Smith-Lemli-Opitz syndrome in order to increase awareness among psychiatrists of this single-gene disorder that is highly associated with autism spectrum disorder. This child illustrates the importance of performing biochemical analyses for Smith-Lemli-Opitz syndrome in individuals with autism spectrum disorder who have specific physical dysmorphology or characteristic behavioral phenotypic features of Smith-Lemli-Opitz syndrome. Most of the parents of children with Smith-Lemli-Opitz syndrome seek help with their children’s behavioral problems, such as irritability, hyperactivity, impulsivity, sleep disturbances, and autistic-like behaviors. Therefore, a psychiatrist may be the first specialist to encounter individuals with this genetic disorder and to establish the diagnosis of Smith-Lemli-Opitz syndrome. Such recognition and understanding will help clinicians implement syndrome-specific treatments of patients identified with a genetic cause of autism spectrum disorder. Early identification of this genetic disorder is critical not only to the individual patient but for the entire family. In addition, the further study of Smith-Lemli-Opitz syndrome and abnormal cholesterol synthesis might lead to the discovery of mechanisms of more general importance to research and treatment of autism spectrum disorder.