Objective: White matter abnormalities may interfere with limbic cortical balance and lead to chronic depressive syndromes. The authors used diffusion tensor imaging to test the hypothesis that depressed elders who fail to achieve remission have microstructural white matter abnormalities in cortico-striato-limbic networks implicated in geriatric depression. Method: The subjects were nondemented individuals with nonpsychotic major depression. After a 2-week placebo period, those subjects who had a Hamilton Depression Rating Scale (HAM-D) score of 18 or greater received escitalopram, 10 mg daily, for 12 weeks. Remission was defined as a HAM-D score of 7 or below for 2 consecutive weeks. Diffusion tensor imaging was performed at a 1.5 Tesla scanner, and voxel-based analysis of fractional anisotropy was conducted using age as the covariate. Results: Subjects who failed to achieve remission (N=23) had lower fractional anisotropy in multiple frontal limbic brain areas, including the rostral and dorsal anterior cingulate, dorsolateral prefrontal cortex, genu of the corpus callosum, white matter adjacent to the hippocampus, multiple posterior cingulate cortex regions, and insular white matter, relative to those who achieved remission (N=25). In addition, lower fractional anisotropy was detected in the neostriatum and midbrain as well as select temporal and parietal regions. Conclusions: Lower fractional anisotropy in distributed cerebral networks is associated with poor antidepressant response of geriatric depression and may represent a neuroanatomical substrate that predisposes to this disorder.
Despite treatment advances, many depressed older adults fail to achieve remission. Depressed patients who achieve remission are three times less likely to relapse than depressed patients who are left with residual symptoms (1) . Moreover, patients who fail to remit have functional impairment and compromised quality of life (2) . Aging-related brain abnormalities may prevent remission by interfering with the regulation of networks needed for normalization of processes contributing to depression.
A confluence of functional imaging findings suggest that depression is associated with abnormal metabolism (mostly increased) in limbic regions, including the amygdala (3), pregenual and subgenual anterior cingulate (4), and posterior orbitofrontal cortex as well as the posterior cingulate and medial cerebellum (5) . In contrast, the lateral and dorsolateral prefrontal cortex, dorsal anterior cingulate, and caudate nucleus have reduced blood flow during depression (6, 7) . Reduced bilateral activation of the dorsal anterior cingulate and the hippocampus has been documented in severely depressed, nondemented elderly patients performing a word activation task (8) . Decreased prefrontal activation was observed in geriatric depressed patients relative to comparison subjects, along with increased caudate activation, during explicit learning of a sequence (9) .
Reciprocal pathways linking midline limbic structures with brainstem, striatal, paralimbic, and neocortical sites have been identified (10) and have contributed to the understanding of the mechanisms of diverse treatment modalities. Depressive symptoms can be reduced by cognitive therapy, a “top down” cortical influence on limbic pathways (11), as well as with pharmacological therapies, which have a “bottom-up” (or combined bottom-up and top-down) effect, since their principal sites of action are the dorsal raphe and locus coeruleus (12) . It has been proposed that abnormalities in the reciprocal relationship between ventral limbic and dorsal cortical systems are critical physiological disturbances in depression (6, 7) . White matter abnormalities compromising connections between limbic and dorsal structures may interfere with limbic cortical balance and contribute to the persistence of depression.
Diffusion tensor imaging may offer information about the structural integrity of white matter areas relevant to the remission of depression. Diffusion tensor imaging measures self-diffusion of water in tissues. When no barriers exist, diffusion occurs equally in all directions (i.e., it is isotropic). However, when barriers to diffusion are present, diffusion follows the long axis of those barriers (i.e., it is anisotropic). The degree to which diffusion is anisotropic depends on microstructural components such as cell membranes and organization of fiber tracts. A measure of anisotropic diffusion is fractional anisotropy. Fractional anisotropy changes occur with aging, cognitive dysfunction, and psychopathology (13) .
The principal assumption of the present study is that microstructural white matter abnormalities may interfere with communication between limbic and dorsal cortical structures, thereby perpetuating depressive symptoms by compromising the aging brain’s ability to re-equilibrate pathological activation that occurs during depressive states (14) . Accordingly, we hypothesized that depressed elders who failed to achieve remission during treatment with escitalopram would have lower fractional anisotropy in areas of cortico-striato-limbic networks implicated in geriatric depression.
Method
Subjects
Subjects were consecutively recruited elderly individuals, age 60 years and older, who agreed to participate in the study and signed informed consent. At entry, subjects met DSM-IV criteria for unipolar major depression without psychotic features and had a score of 18 or greater on the 24-item Hamilton Depression Rating Scale (HAM-D).
Depressed patients were excluded if they had 1) history of other psychiatric disorders (except personality disorders) prior to the onset of depression; 2) severe medical illness (i.e., metastatic cancer; brain tumors; unstable cardiac, hepatic, or renal disease; myocardial infarction; or stroke) within the 3 months preceding the study; 3) neurological disorders (i.e., delirium, history of stroke, head trauma, multiple sclerosis, and brain degenerative diseases, including Parkinson’s disease); 4) conditions or use of drugs that may cause depression (i.e., endocrinopathies other than diabetes, lymphoma, pancreatic cancer or treatment with steroids, alpha-methyl-dopa, clonidine, reserpine, tamoxifen, and cimetidine); and 5) Mini-Mental State Examination score <24. Therefore, the subjects were nondemented, nondelusional elderly patients with major depression.
Measures
The Structured Clinical Interview for DSM-III-R (SCID-R) was initially administered when the subjects were symptomatic. Depressive symptoms were assessed using the 24-item HAM-D. Side effects of escitalopram were monitored with the UKU Side Effect Rating Scale (15) . Baseline cognitive impairment was rated with the Mini-Mental State Examination, and disability was rated with the World Health Organization Disability Assessment Schedule (16) .
Treatment
Subjects were informed that they would receive placebo at some point during the 14-week trial. After a 2-week drug washout and single-blind placebo lead-in, subjects who still met DSM-IV criteria for major depression and had a HAM-D score of 18 or greater received controlled treatment with escitalopram, 10 mg daily, for 12 weeks. Escitalopram was given in a single dose in the morning. Subjects received their medication in 1-week supply blisters, which permitted dispensation of their daily dosage separately.
During the treatment phase, the subjects were followed weekly from the time escitalopram was started until week 12. Each follow-up meeting consisted of a rating session, with a research assistant who administered the HAM-D and UKU Side Effect Rating Scale, obtained vital signs, questioned subjects about medication adherence, and counted the remaining tablets. The meeting with the research assistant was followed by a brief session with a research psychiatrist, the purpose of which was to assess the risk of continuing the treatment trial. This session followed a medication clinic model consisting of a review of symptoms, explanations related to the need for treatment, and encouragement of treatment adherence. No subject received psychotherapy during the study. The subjects were considered to be in remission if they no longer met DSM-IV criteria for depression and had a HAM-D score of 7 or below for 2 consecutive weeks.
Magnetic Resonance Imaging
Scans were obtained with the 1.5T Siemens Vision Scanner (Erlangen, Germany) housed at the Nathan Kline Institute (NKI) Center for Advanced Brain Imaging. Patients received a magnetization-prepared rapidly acquired gradient echo (MPRAGE) scan (TR=11.6 msec, TE=4.9 msec, matrix=256×256, field of view=320 mm, number of excitations=1, slice thickness=1.25 mm, 172 slices, no gap) as well as a turbo dual-spin echo (TSE) scan (TR=5000 msec, TE=22/90 msec, matrix=256×256, field of view=240 mm, slice thickness=5 mm, 26 slices, no gap) and a diffusion tensor imaging scan (TR=6000 msec, TE=100 msec, matrix=128×128, field of view=320 mm, number of excitations=7, slice thickness=5 mm, 19 slices, no gap). For the diffusion tensor imaging scan, eight diffusion sensitization directions were used (b=1000 seconds/mm 2 ), along with an image with no diffusion weighting (b=0 seconds/mm 2 ). The TSE and diffusion tensor imaging scans were acquired in an oblique axial plane parallel to the anterior commissure–posterior commissure axis.
Fractional anisotropy was computed using AFNI nonlinear algorithm (3dDWItoDT). Diffusion tensor images were placed into Talairach space using methods described elsewhere (17) . Intersubject registration was conducted using an automatic registration toolkit (18) . The late echo of the TSE scan was used to correct for susceptibility-induced distortion.
The T1-weighted template used for the registrations was derived from a subject whose intracranial content volume was the closest to the mean for the first five subjects. The volumes were computed in MEDx (Sensor Systems, Sterling, Va.) after skull stripping, which was conducted using an FMRIB Software Library brain extraction tool program (http://www.fmrib.ox.ac.uk/analysis/research/bet/). The case we selected was placed into Talairach space using AFNI (19) .
Images were masked for white matter by computing the mean fractional anisotropy map from a larger group of 83 patients, which included the 48 patients reported in this study, and performing a nonparametric histogram-based segmentation. The obtained white matter threshold was applied to the mean fractional anisotropy map, and the resulting mask was applied to each subject’s normalized fractional anisotropy.
Voxel-based analysis of fractional anisotropy data, with respect to group membership (remitting, nonremitting), was conducted using a general linear model with age as the covariate. To control for type I error, we used the thresholding method described by Baudewig et al. (20), which we also used in a previous study (17) . This method locates clusters of contiguous voxels (100 mm 3 ) with significant group differences (p<0.05) and then applies the constraint so that one of the voxels in the cluster must be significant at p<0.005. The thresholded correlation maps were superimposed onto a magnetization-prepared rapidly acquired gradient echo image in Talairach space using AFNI (19) .
Results
A total of 62 subjects met selection criteria and entered the 2-week single-blind placebo phase. Of these 62 subjects, 48 continued to meet symptom severity criteria (HAM-D score: 18–34; mean=22.2) after the 2-week placebo phase and entered the 12-week escitalopram treatment phase. Of these 48 subjects, 39 completed the 12-week treatment trial. Of the remaining nine subjects, three had 4 weeks of treatment (all exited because of worsening depression), two had 7 weeks of treatment (one exited because he found the treatment ineffective, and one withdrew because she developed hyponatremia), one had 8 weeks of treatment (and exited because he found the treatment ineffective), one had 9 weeks of treatment (and exited because of worsening depression), and two had 11 weeks of treatment with escitalopram (both exited because they found the treatment ineffective). All subjects, with the exception of one, who exited prior to 12 weeks failed to achieve remission.
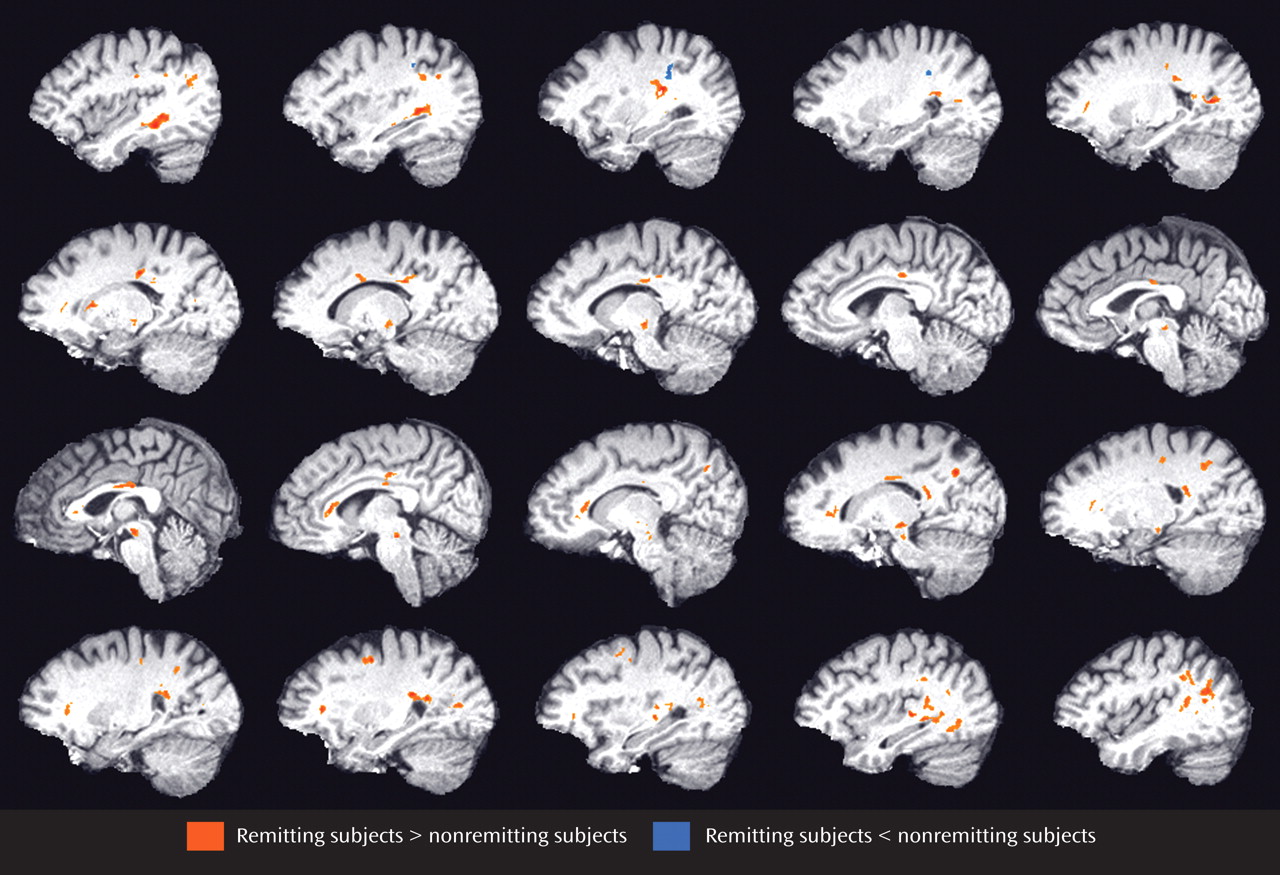
There were no significant differences in age, gender, education, age at first onset of depression, number of previous episodes, severity of depression, or cognitive impairment at baseline between subjects who achieved remission and those who remained symptomatic ( Table 1 ). At study exit, subjects who achieved remission had lower HAM-D scores than those who did not achieve remission (mean: 4.7 [SD=4.4)] versus 17.1 [SD=7.3], t=7.0, df=35.6, p<0.0001).
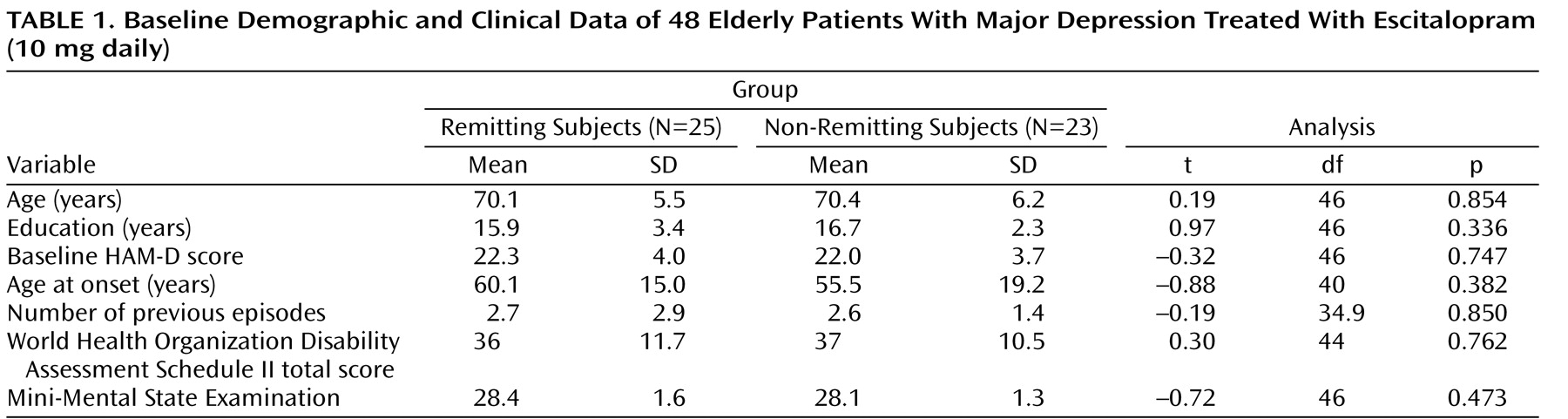
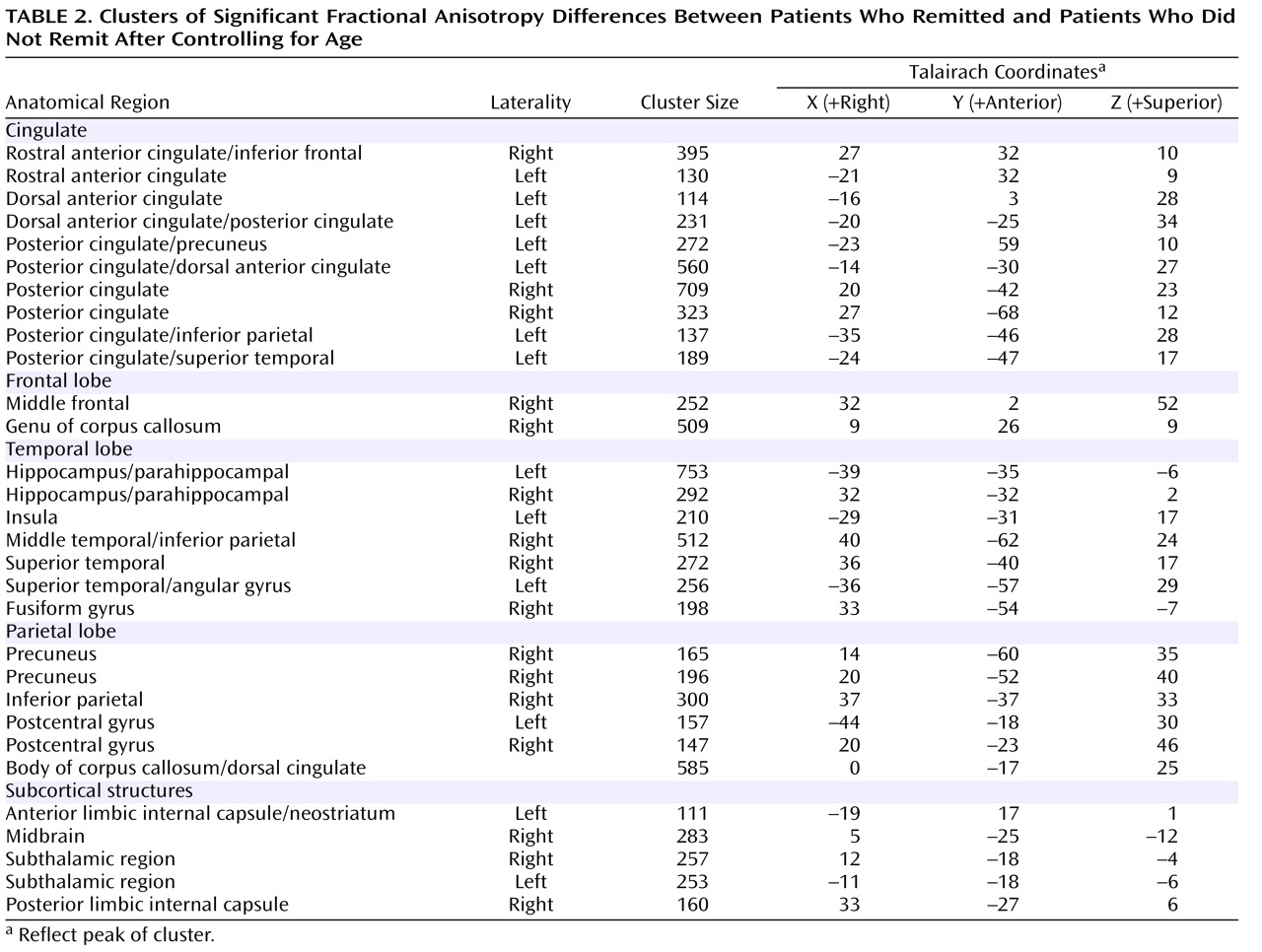
Relative to those subjects who achieved remission (N=25), subjects who failed to achieve remission (N=23) had lower fractional anisotropy in multiple frontal limbic brain areas, including the rostral and dorsal anterior cingulate, dorsolateral prefrontal cortex, genu of the corpus callosum, white matter adjacent to the hippocampus, multiple posterior cingulate regions, and insular white matter ( Figure 1 ). Lower fractional anisotropy was also detected in the neostriatum and subcortical white matter regions. In addition, select temporal (superior, middle, and fusiform) and parietal (precuneus, inferior parietal, and postcentral) white matter regions demonstrated lower fractional anisotropy values in subjects who failed to achieve remission. In nonremitting subjects, two clusters of higher fractional anisotropy were present in the left precentral gyrus and left inferior parietal white matter ( Figure 1, Table 2 ).
Figure 1. Significant Differences in Fractional Anisotropy Between Remitting and Nonremitting Subjects a
a Data from sagittal slices are shown from left to right hemisphere. Group differences have been thresholded so that 100-mm 3 clusters of contiguous voxels significant at p<0.05 are identified, with the additional criterion that at least one voxel is significant at p<0.005 within each cluster. The figure shows a high-resolution T1-weighted template, with age as a covariate.
The principal finding of this study is that depressed elderly patients who remained symptomatic after treatment with escitalopram had lower fractional anisotropy in several cortico-striato-limbic white matter areas relative to depressed elders who achieved remission. With the exception of our preliminary report (21), this is the first study, to our knowledge, identifying a relationship between reduced fractional anisotropy across various brain areas and remission of geriatric depression.
Structural neuroimaging revealed white matter abnormalities in late-life depression. White matter hyperintensities are common in elderly depressed individuals and mainly occur in subcortical regions and their frontal white matter projections (22) . Diffusion tensor imaging revealed that depressed elders have compromised white matter integrity at the anterior cingulate cortex and select frontal and temporal regions (23, 24) . Magnetization transfer imaging, a technique used to assess macromolecular integrity, showed abnormalities in geriatric depression in fronto-striato-thalamo-limbic networks and structures connected to these networks, including the neostriatum and occipital white matter and the genu and splenium of the corpus callosum (25) .
Our findings are consistent with observations suggesting that some structural brain abnormalities are associated with poor outcomes of late-life depression. White matter hyperintensities have been associated with chronicity of geriatric depression (26), although disagreement exists (27) . In one study, basal ganglia lesions predicted failure to respond to antidepressant monotherapy, with a sensitivity of 80% and a specificity of 62% (28) . In another study, lesions in subcortical gray matter predicted poor outcome of geriatric depression over a period extending to 64 months (29) . Finally, a preliminary diffusion tensor imaging study noted that reduced fractional anisotropy in white matter lateral to the anterior cingulate cortex predicted poor remission rates among elderly patients with major depression treated with citalopram (21) .
Functional neuroimaging has also shown that improvement of depression is associated with cortico-striato-limbic changes. Electromagnetic tomography in adults with major depression revealed that hyperactivity in the rostral anterior cingulate cortex (Brodmann’s areas 24/32) predicted favorable response to nortriptyline (30) . Remission of depression is associated with metabolic increases in dorsal cortical regions (dorsal anterior cingulate cortex [Brodmann’s area 24b], posterior cingulate cortex [Brodmann’s areas 23/41], dorsolateral cortex [Brodmann’s areas 46/9], and inferior parietal cortex [Brodmann’s area 40]) (6, 31, 32) . In contrast, decreases in ventral limbic and paralimbic structures (subgenual cingulate cortex [Brodmann’s area 25]), ventral mid- and posterior insula, hippocampus, and hypothalamus) occur during remission (32) . Remission of depression is associated with normalization of anterior cingulate cortex metabolic abnormalities that occur during depressive episodes (5, 32) . Rostral anterior cingulate cortex hypermetabolism subsides after response to antidepressants (33), electroconvulsive therapy (34), or sleep deprivation (35), although the normalization pattern depends on the treatment modality (selective serotonin reuptake inhibitor versus cognitive behavior therapy) (36) . Persistently elevated amygdala metabolism during remission of depression is associated with high risk for relapse of depression (3) . Hypometabolism of the rostral anterior cingulate cortex is a predictor of treatment resistance in younger depressed patients, while cingulate hypermetabolism predicts favorable response (33) . Finally, increased metabolism of the left anterior cingulate cortex was found in young, depressed, poor treatment responders (37) .
These observations suggest that metabolic changes occur during depression in cortico-striato-limbic systems and improvement of depression is associated with the restoration of some of these changes. A potential explanation of our findings is that some of the observed microstructural abnormalities in cortico-striato-limbic networks interfere with the reciprocal regulation of dorsal, neocortical, ventral limbic structures and lead to a “disconnection syndrome” associated with poor antidepressant response. Combining structural and functional imaging methods may help in distinguishing those microanatomical abnormalities that are predisposing to nonremission of geriatric depression from incidental abnormalities.
The findings of this study should be viewed in the context of the following limitations: the subject selection bias inherent in imaging studies (healthier patients may be preferentially included), lack of a placebo comparison group, fixed dose of the antidepressant, limited assessment of cognitive functions, and absence of long-term follow-up. It is likely that some subjects may have remitted if they had been treated with higher doses of escitalopram, if the dose of escitalopram had been increased at week 5 to week 6 of treatment in those subjects who failed to improve, or if a longer treatment period had been offered. However, six out of nine nonremitting subjects who exited the trial had 7 to 11 weeks of escitalopram treatment, and three had 4 weeks of treatment. Moreover, there were no significant differences at baseline between remitting and nonremitting subjects in variables associated with prediction of treatment resistance. Finally, our observations may be influenced by the large number of comparisons, since voxelwise analysis increases the risk of type I error. To address this potential problem, we used a white matter mask to reduce the number of comparisons. We also applied a rather conservative threshold and cluster size requirement for additional protection over type I error.
The etiology of microanatomical abnormalities of elderly nonremitting subjects is unclear. Vascular changes preferentially damage subcortical structures. The hippocampus is particularly vulnerable to aging and aging-related changes. Reduced hippocampal volume is correlated with the lifetime duration of depression (38) . Moreover, decline in hippocampal volume has been documented after the first episode of major depression, even in patients receiving antidepressant treatment. Finally, abnormalities in each of these structures occur early in Alzheimer’s disease. Reduced metabolism in the posterior cingulate cortex and precuneus has been reported in presymptomatic nondemented individuals who had at least a single apolipoprotein epsilon 4 allele (39) . Similarly, decreased metabolism in the posterior cingulate cortex, hippocampus, and temporoparietal association areas was reported in patients with mild cognitive impairment who converted to dementia (40) . Thus, one or more factors may contribute to the microanatomical abnormalities that are predisposing to resistance to antidepressants in late-life depression.
This study suggests that white matter abnormalities in cortico-striato-limbic networks confer vulnerability increasing the likelihood of chronicity of geriatric depression. Viewing these abnormalities as predisposing rather than causing chronicity of depression may explain the effect of other clinical (e.g., history of multiple episodes) and nonclinical factors (e.g., chronic adversity) on the course of geriatric depression. These observations can be used as the basis for brain function studies aiming to identify impairment in specific fronto-striato-limbic functions that interfere with the response of geriatric depression to antidepressants.
Footnotes
Received May 3, 2007; revisions received June 13, 2007; accepted July 3, 2007 (doi: 10.1176/appi.ajp.2007.07050744). From Weill Cornell Institute of Geriatric Psychiatry, Weill Cornell Medical College, New York; Nathan S. Kline Institute for Psychiatric Research, Orangeburg, N.Y.; Department of Psychiatry, New York University School of Medicine, New York; and University of Minnesota, Minneapolis. Address correspondence and reprint requests to Dr. Alexopoulos, 21 Bloomingdale Rd., White Plains, NY 10605; gsalexop@med. cornell.edu (e-mail).
Dr. Alexopoulos has received research grant funding from Forest Pharmaceuticals and Cephalon; he has participated in scientific advisory board meetings for Forest Pharmaceuticals; he has given lectures supported by Forest, Cephalon, Bristol-Meyers Squibb, GlaxoSmithKline, Pfizer, Janssen, and Lilly; and he has received support from Comprehensive Neuroscience for development of treatment guidelines in late-life psychiatric disorders. Drs. Murphy, Gunning-Dixon, Latoussakis, Klimstra, Lim, and Hoptman and Ms. Kanellopoulos report no competing interests.
Supported by NIMH grants R01 MH65653, P030 MH68638, T32 MH019132 (Dr. Alexopoulos), K23 MH067702 (Dr. Murphy), K23 MH74818 (Dr. Gunning-Dixon), the Sanchez Foundation, and Forest Pharmaceuticals.
The authors thank Raj Sangoi, R.T.(R)(MR) for his work as chief MR technologist.
References
1.
Judd LL, Akiskal HS, Maser JD, Zeller PJ, Endicott J, Coryell W, Paulus MP, Kunovac JL, Leon AC, Mueller TI, Rice JA, Keller MB: Major depressive disorder: a prospective study of residual subthreshold depressive symptoms as predictor of rapid relapse. J Affect Disord 1998; 50:97–108
Buchsbaum MS, Wu J, Siegel BV, Hackett E, Trenary M, Abel L, Reynolds C: Effect of sertraline on regional metabolic rate in patients with affective disorder. Biol Psychiatry 1997; 41:15–22
Mayberg H: Depression and frontal-subcortical circuits: focus on prefrontal-limbic interactions, in Frontal-Subcortical Circuits in Psychiatric and Neurological Disorders. Edited by Lichter D, Cummings J. New York, Guilford Press, 2001, pp 177–206
de Asis JM, Stern E, Alexopoulos GS, Pan H, Van Gorp W, Blumberg H, Kalayam B, Eidelberg D, Kiosses D, Silbersweig DA: Hippocampal and anterior cingulate activation deficits in patients with geriatric depression. Am J Psychiatry 2001; 158:1321–1323
Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K: The UKU Side Effect Rating Scale: a new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr Scand Suppl 1987; 334:1–100
Taylor WD, MacFall JR, Payne ME, McQuoid DR, Steffens DC, Provenzale JM, Krishnan RR: Greater MRI lesion volumes in elderly depressed subjects than in control subjects. Psychiatry Res 2005; 139:1–7
Kumar A, Gupta RC, Albert Thomas M, Alger J, Wyckoff N, Hwang S: Biophysical changes in normal-appearing white matter and subcortical nuclei in late-life major depression detected using magnetization transfer. Psychiatry Res 2004; 130:131–140
Hickie I, Scott E, Mitchell P, Wilhelm K, Austin M, Bennett B: Subcortical hyperintensities on magnetic resonance imaging: clinical correlates and prognostic significance in patients with severe depression. Biol Psychiatry 1995; 37:151–160
Salloway S, Boyle P, Correia S, Malloy P, Cahn-Weiner D, Schneider L, Krishnan K, Nakra R: The relationship of MRI subcortical hyperintensities to treatment response in a trial of sertraline in geriatric depressed outpatients. Am J Geriatr Psychiatry 2002; 10:107–111
Patankar TF, Baldwin R, Mitra D, Jeffries S, Sutcliffe C, Burns A, Jackson A: Virchow-robin space dilatation may predict resistance to antidepressant monotherapy in elderly patients with depression. J Affect Disord 2007; 97:265–270
Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, Silva JA, Tekell JL, Martin CC, Lancaster JL, Fox PT: Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry 1999; 156:675–682
Nobler MS, Oquendo MA, Kegeles LS, Malone KM, Campbell CC, Sackeim HA, Mann JJ: Decreased regional brain metabolism after ECT. Am J Psychiatry 2001; 158:305–308
Goldapple K, Segal Z, Garson C, Lau M, Bieling P, Kennedy S, Mayberg H: Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry 2004; 61:34–41
Schneider F, Grodd W, Weiss U, Klose U, Mayer K, Nagele T, Gur R: Functional MRI reveals left amygdala activation during emotion. Psychiatry Res 1997; 76:75–82
Sheline Y: 3D MRI studies of neuroanatomic changes in unipolar major depression: the role of stress and medical comorbidity. Biol Psychiatry 2000; 48:791–800
Wolf H, Jelic V, Gertz HJ, Nordberg A, Julin P, Wahlund LO: A critical discussion of the role of neuroimaging in mild cognitive impairment. Acta Neurol Scand Suppl 2003; 179:52–76
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
Figure 1. Significant Differences in Fractional Anisotropy Between Remitting and Nonremitting Subjects a
a Data from sagittal slices are shown from left to right hemisphere. Group differences have been thresholded so that 100-mm 3 clusters of contiguous voxels significant at p<0.05 are identified, with the additional criterion that at least one voxel is significant at p<0.005 within each cluster. The figure shows a high-resolution T1-weighted template, with age as a covariate.
Buchsbaum MS, Wu J, Siegel BV, Hackett E, Trenary M, Abel L, Reynolds C: Effect of sertraline on regional metabolic rate in patients with affective disorder. Biol Psychiatry 1997; 41:15–22
Mayberg H: Depression and frontal-subcortical circuits: focus on prefrontal-limbic interactions, in Frontal-Subcortical Circuits in Psychiatric and Neurological Disorders. Edited by Lichter D, Cummings J. New York, Guilford Press, 2001, pp 177–206
de Asis JM, Stern E, Alexopoulos GS, Pan H, Van Gorp W, Blumberg H, Kalayam B, Eidelberg D, Kiosses D, Silbersweig DA: Hippocampal and anterior cingulate activation deficits in patients with geriatric depression. Am J Psychiatry 2001; 158:1321–1323
Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K: The UKU Side Effect Rating Scale: a new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr Scand Suppl 1987; 334:1–100
Taylor WD, MacFall JR, Payne ME, McQuoid DR, Steffens DC, Provenzale JM, Krishnan RR: Greater MRI lesion volumes in elderly depressed subjects than in control subjects. Psychiatry Res 2005; 139:1–7
Kumar A, Gupta RC, Albert Thomas M, Alger J, Wyckoff N, Hwang S: Biophysical changes in normal-appearing white matter and subcortical nuclei in late-life major depression detected using magnetization transfer. Psychiatry Res 2004; 130:131–140
Hickie I, Scott E, Mitchell P, Wilhelm K, Austin M, Bennett B: Subcortical hyperintensities on magnetic resonance imaging: clinical correlates and prognostic significance in patients with severe depression. Biol Psychiatry 1995; 37:151–160
Salloway S, Boyle P, Correia S, Malloy P, Cahn-Weiner D, Schneider L, Krishnan K, Nakra R: The relationship of MRI subcortical hyperintensities to treatment response in a trial of sertraline in geriatric depressed outpatients. Am J Geriatr Psychiatry 2002; 10:107–111
Patankar TF, Baldwin R, Mitra D, Jeffries S, Sutcliffe C, Burns A, Jackson A: Virchow-robin space dilatation may predict resistance to antidepressant monotherapy in elderly patients with depression. J Affect Disord 2007; 97:265–270
Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, Silva JA, Tekell JL, Martin CC, Lancaster JL, Fox PT: Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry 1999; 156:675–682
Nobler MS, Oquendo MA, Kegeles LS, Malone KM, Campbell CC, Sackeim HA, Mann JJ: Decreased regional brain metabolism after ECT. Am J Psychiatry 2001; 158:305–308
Goldapple K, Segal Z, Garson C, Lau M, Bieling P, Kennedy S, Mayberg H: Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry 2004; 61:34–41
Schneider F, Grodd W, Weiss U, Klose U, Mayer K, Nagele T, Gur R: Functional MRI reveals left amygdala activation during emotion. Psychiatry Res 1997; 76:75–82
Sheline Y: 3D MRI studies of neuroanatomic changes in unipolar major depression: the role of stress and medical comorbidity. Biol Psychiatry 2000; 48:791–800
Wolf H, Jelic V, Gertz HJ, Nordberg A, Julin P, Wahlund LO: A critical discussion of the role of neuroimaging in mild cognitive impairment. Acta Neurol Scand Suppl 2003; 179:52–76
Figure 1. Significant Differences in Fractional Anisotropy Between Remitting and Nonremitting Subjects a
a Data from sagittal slices are shown from left to right hemisphere. Group differences have been thresholded so that 100-mm 3 clusters of contiguous voxels significant at p<0.05 are identified, with the additional criterion that at least one voxel is significant at p<0.005 within each cluster. The figure shows a high-resolution T1-weighted template, with age as a covariate.
Table 1
No caption available.
Table 2
No caption available.
Reference #1
Request Username
If the address matches an existing account you will receive an email with instructions to retrieve your username
Create a new account
Change Password
Password Changed Successfully
Your password has been changed
Login
Reset password
Can't sign in? Forgot your password?
Enter your email address below and we will send you the reset instructions
If the address matches an existing account you will receive an email with instructions to reset your password.
Change Password
Congrats!
Your Phone has been verified
×
As described within the American Psychiatric Association (APA)'s Privacy Policy and Terms of Use, this website utilizes cookies, including for the purpose of offering an optimal online experience and services tailored to your preferences. Please read the entire Privacy Policy and Terms of Use. By closing this message, browsing this website, continuing the navigation, or otherwise continuing to use the APA's websites, you confirm that you understand and accept the terms of the Privacy Policy and Terms of Use, including the utilization of cookies.