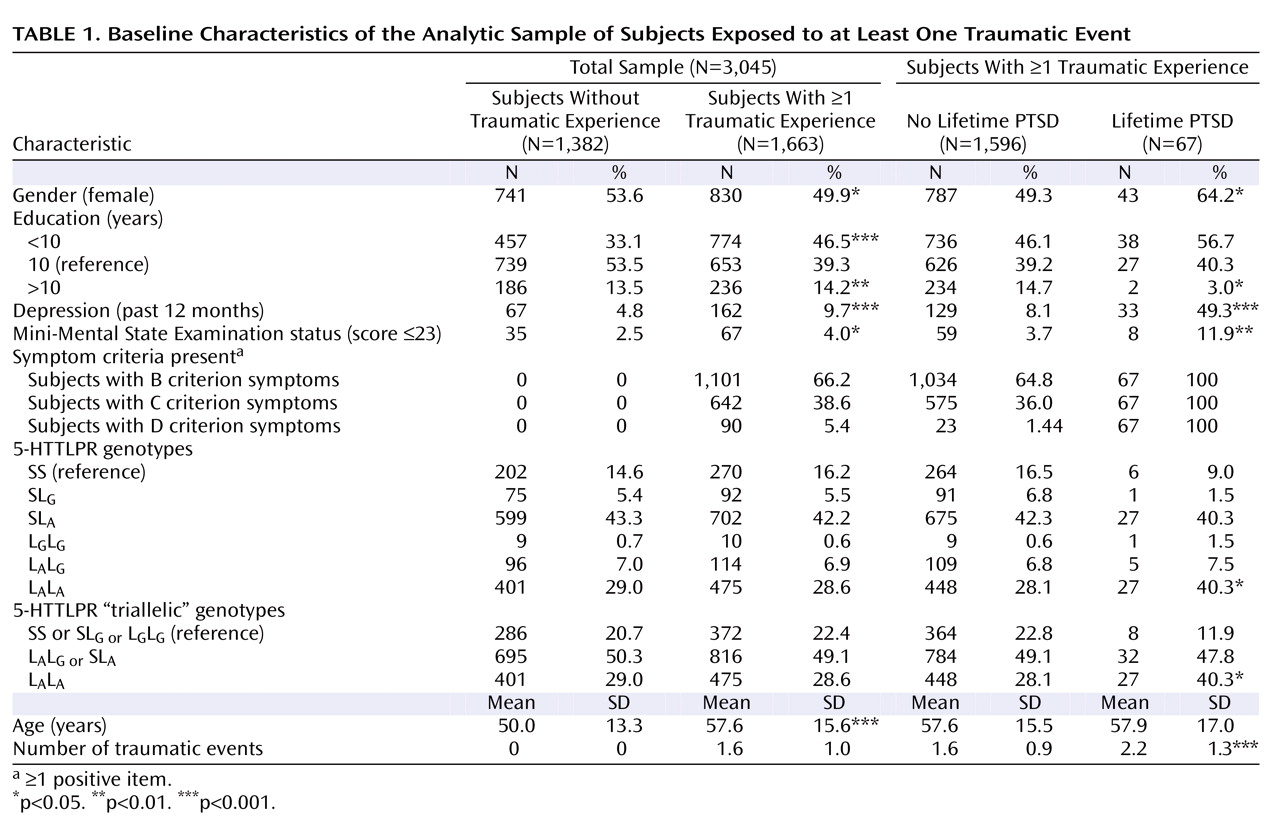
The description of clinical and demographic characteristics of the sample is provided in
Table 1 . There was no departure from the Hardy-Weinberg equilibrium (total sample [triallelic]: χ
2 =2.53, df=2, p=0.28; exposed sample [triallelic]: χ
2 =1.18, df=2, p=0.55). All genetic analyses were based on 1,663 subjects who reported exposure to at least one traumatic event. Sixty-seven subjects (4.03%) met the diagnostic criteria for lifetime diagnosis of PTSD. There were no associations between the number of traumatic events and the triallelic genotypes (F=0.115, df=2, 1660, p=0.89).
Gene Main Effects
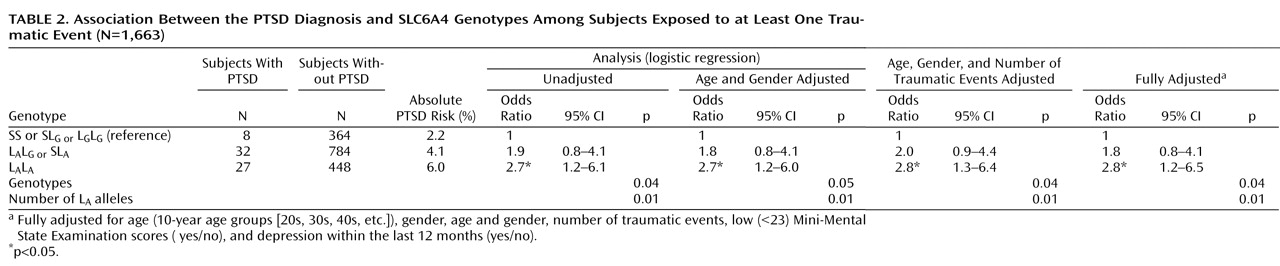
Main effects of the three genotypes with the diagnosis of PTSD (L
A /L
A ; L
G /L
A or S/L
A ; and L
G /L
G or L
G /S or S/S) were analyzed by logistic regression analysis (
Table 2 ). All associations between the L
A /L
A genotype and PTSD (both crude and adjusted) were statistically significant. There was clear evidence for a dose-response relationship, since among subjects with zero, one, and two L
A alleles the crude proportion of PTSD increased from 2% to 4% and 6%, respectively. The Cochran-Armitage test indicated a clear additive mode for L
A alleles (χ
2 =6.78, df=1, p=0.009), with no departure from linearity (χ
2 =0.00, df=1, p=0.99). The allelic analyses revealed an effect for the L
A allele (odds ratio=1.6; 95% CI=1.1–2.4) but not for the more rare L
G allele (odds ratio=0.7; 95% CI=0.3–1.6) on PTSD in the fully adjusted model (see the data supplement accompanying the online version of this article). No associations were established between the 5-HTTLPR genotypes and criteria B, C, or D (p≥0.05).
Gene-Environment Interaction
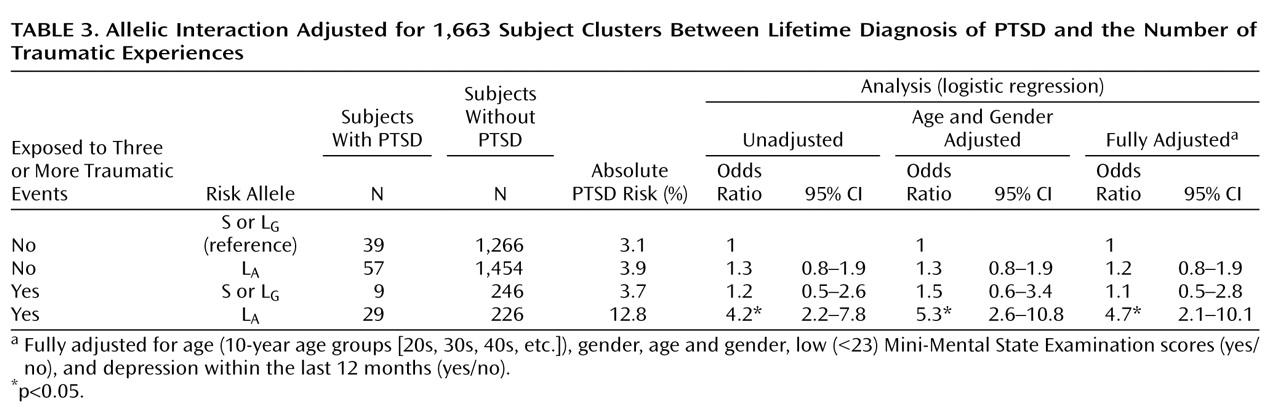
In allelic analyses, the estimated absolute risk among subjects who were not exposed to three or more traumatic events was 3.1% for those without the L
A allele and 3.9% for those with the L
A allele; whereas the risk among subjects who were exposed to three or more traumatic events was 3.7% for those without the L
A allele but 12.8% for those with the L
A allele (
Table 3 ). Thus, based on the same difference in absolute risks for the L
A allele in trauma-exposed and nonexposed subjects, the absolute additive excess risk for trauma-exposed subjects with the L
A allele was as follows: 12.8%–3.7%–3.9%+3.1%=8.3%. The unadjusted RERI was then 8.3%/3.1%=2.7, which is in terms of the odds ratio equivalent to 2.7=4.2–1.2–1.3+1 (
Table 3 ). Thus, the unadjusted odds ratio of having the lifetime diagnosis of PTSD among subjects with ≥3 traumas and the L
A allele relative to those without the L
A allele was 2.7 times higher than the condition in which no interaction between traumas and the L
A allele was present (e.g., absolute risk of 4.5% instead of 12.8% or, equivalently, odds ratio=4.5/3.1=1.5 instead of odds ratio=4.2). For the final model, RERI was 3.3 (95% CI=1.2–8.2 [
Table 4 ]). Importantly, RERI–1>0 holds for the 95% CI. The unadjusted attributable proportion was 2.7/4.2=0.64 (64%), and the fully adjusted attributable proportion was 71% (95% CI=41–88), meaning that approximately 71% of PTSD diagnoses among subjects with ≥3 traumas and the L
A allele was attributable to the interaction between the two.
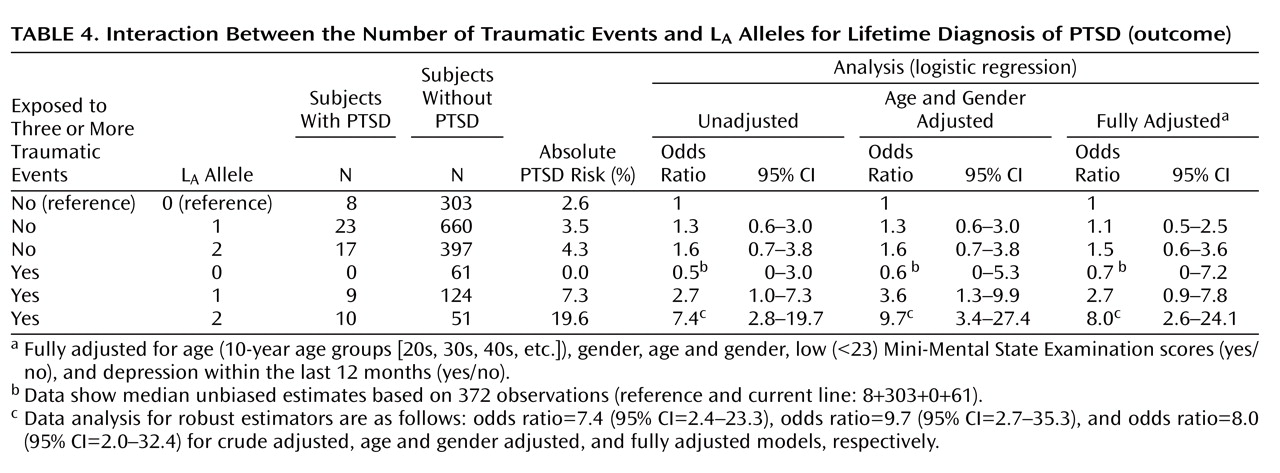
To avoid a sparse data problem, referring to a genotype-trauma combination with no cases of PTSD (
Table 4 ), subjects with zero or one L
A allele were grouped into one category versus subjects with two L
A alleles. The absolute risks were 31/963=3.2% and 4.3% among nonexposed subjects without and with two L
A alleles, respectively, and 9/185=4.9% and 19.6% among exposed subjects without and with two L
A alleles, respectively (
Table 4 ). This type of RERI was crude (13.6%/3.2%=4.25) and fully adjusted (5.5) (95% CI=0.8–18.3, p<0.05). The unadjusted attributable proportion was 4.25/(10×963/[31×51])=4.25/6.1=0.70 (70%), and the fully adjusted attributable proportion was 74% (95% CI=18–92).
Since the allelic analyses supported that subjects with only one L
A allele possibly had a higher risk for PTSD when trauma exposed compared with those subjects carrying no L
A alleles, we calculated RERI for the number of L
A alleles as a continuous variable. This type of RERI directly estimates the relative excess risk for one L
A allele compared with no L
A alleles. In the present study, RERI was 1.3 for the unadjusted model and 1.5 (95% CI=0.5–4.3, p<0.05) for the fully adjusted model (
Table 4 ). This means that among subjects with ≥3 traumas the odds ratio of having PTSD was 1.5 times higher with one L
A allele compared with the absence of interaction between the L
A alleles and traumas. We confirmed these results using a Bayesian approach to address the sparse data problem (see the data supplement accompanying the online version of this article). Prior limits similar to the related CIs of the fully adjusted model for the number of symptoms from the D criterion were utilized (see the data supplement accompanying the online version of this article).
When four instead of three was used as a cutoff point for the number of traumas, RERI for the dichotomized genotype (zero or one L A allele versus two L A alleles) increased to 8.8 (95% CI=1.6–31.7, p<0.05) and attributable proportion increased to 86% (95% CI=33–97). Given the lower number of subjects with ≥4 traumatic events, the continuous models were less robust. When two was used as a cutoff point for the number of traumas, no statistically significant interactions emerged.
For the number of symptoms from the D criterion as the outcome (see the data supplement accompanying the online version of this article), no genotype-trauma combination with a zero count occurred. RERI for the continuous genetic model (zero, one, or two L A alleles) was 1.2 (95% CI=0.5–2.8, p<0.05) and attributable proportion for the continuous genetic model was 72% (95% CI=23–126). No statistically significant interactions emerged when performing the analyses for B and C criteria irrespective of the trauma threshold.