Understanding the molecular mechanisms underlying major depressive disorder is essential because one in six individuals in the United States will develop depressive symptoms requiring treatment (
1), depression significantly complicates chronic illness (
2), and depression is the leading cause of disability worldwide (
3). However, exploring the molecular underpinnings of depression brings substantial challenges. In contrast to the clear-cut phenotypes encountered in substance dependence or obesity, the strictest guidelines for diagnosing depression include elements that are difficult to capture in animal models, e.g., “insomnia or hypersomnia nearly every day” (
4). Unlike Parkinson's or Alzheimer's disease, depression lacks any clear consensus neuropathology, rare familial genetic causes, or highly penetrant vulnerability genes, providing no obvious starting points for molecular investigations. In spite of its heritability, the search for genetic causes has not been successful to date. Consequently, progress in understanding the molecular biology of depression has been slow, particularly in comparison to other multifactorial syndromes, such as type 2 diabetes mellitus and cancer. Thus, the burden of depression will continue to increase (
3), especially during the extra years of life gained from improved outcomes in cardiovascular disease, cancer, and other domains.
Nevertheless, it is an exciting time to be a depression researcher. Advances in molecular tools and ongoing improvements in behavioral techniques have allowed for genuinely novel insights into depression's neurobiological correlates. Methods to capture and artificially stimulate or inhibit the electrophysiological activity of individual types of neurons in the brain in vivo have added new dimensions to available approaches, permitting us for the first time to describe and manipulate the previously enigmatic neurophysiological correlates of concepts such as “reward” and “anxiety.” Coupled with experimental advances in treatment, these developments suggest we can anticipate that developing tomorrow's therapies will no longer rely solely on modifications of existing agents that were discovered by serendipity six decades ago. Our aim in this review is to provide a framework to interpret continuing advances in the basic science of depression.
Insights From Human Studies
While animal experiments offer a unique opportunity to test cellular and molecular hypotheses, human clinical investigation continues to provide insights about depression that are inaccessible in animals. Postmortem studies designed to capture the neuropathology of depression have largely focused on certain cortical and hippocampal regions, which show a number of subtle differences, such as smaller neuronal size, fewer glial cells, shorter dendrites, and lower levels of trophic factors (
5–7). These results agree with evidence of volume loss in these regions as shown by structural magnetic resonance imaging (MRI) (
8,
9). Molecular techniques such as DNA microarray profiling have been applied to specific regions, including the amygdala and locus ceruleus, to document gene expression alterations associated with depression (
10,
11). Within the coming years, we can hope for a more comprehensive list of depression's neuropathological changes, particularly with the advent of centralized brain collections, which are able to furnish larger samples while simultaneously excluding traditional sources of confound, such as suicide, comorbid substance abuse, and a bipolar diagnosis. When elegantly combined with animal models or neuroimaging data, these postmortem depression studies provide the opportunity to demonstrate true causal relationships (
12–15).
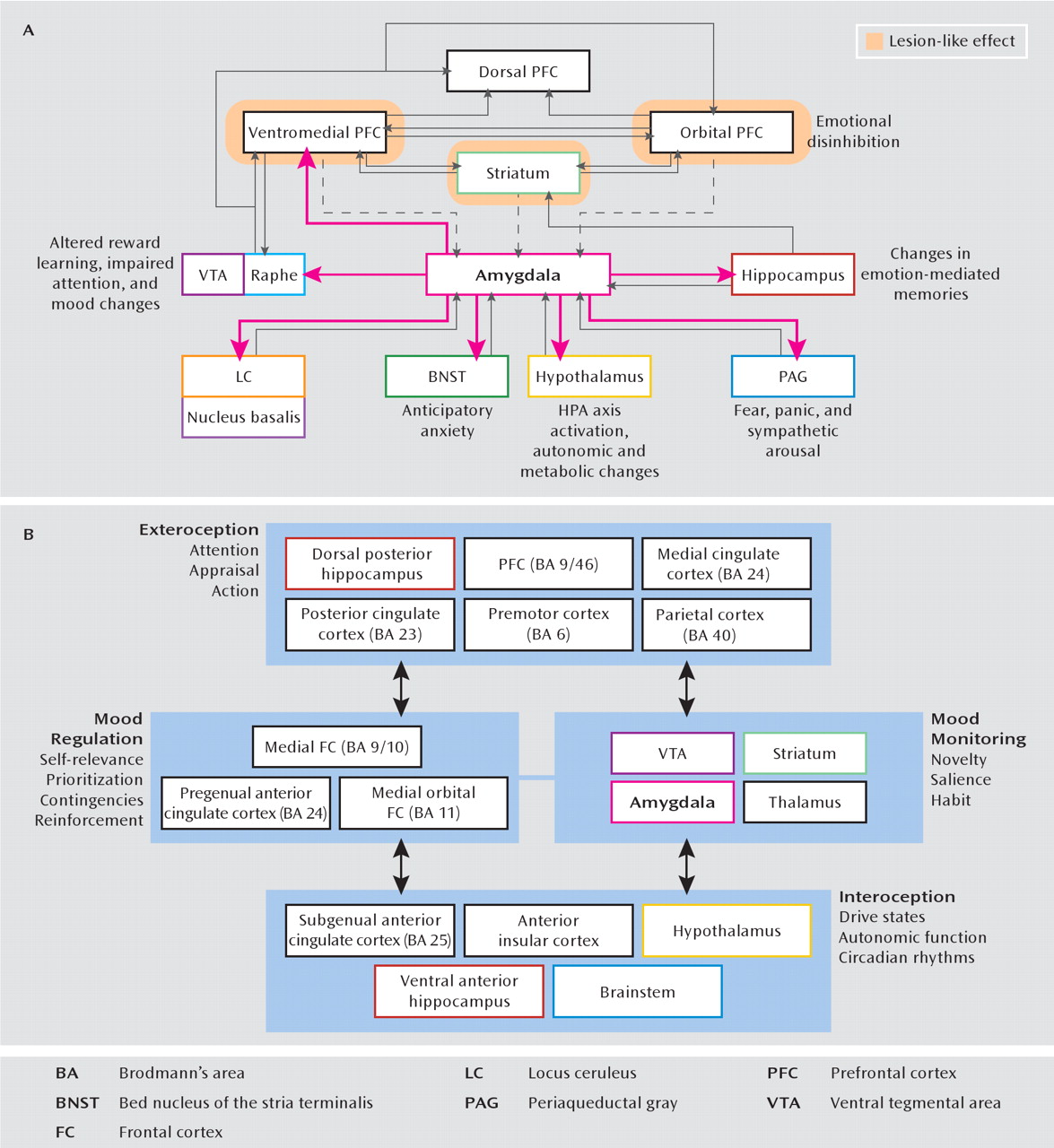
Functional MRI and positron emission tomography (PET) have shown how depressive behavior can be correlated with hypermetabolism of the subgenual cingulate cortex and amygdala (
16) as well as hypometabolism of the dorsal prefrontal cortex and striatal regions (
8). In an attempt to integrate these anatomic data, there have been several formulations of a “depression circuit” (
Figure 1). After years of largely empirical reports, we now approach the possibility of testing and refining these circuit models in humans, thanks to recent experimental interventional advances such as deep brain stimulation and repetitive transcranial magnetic stimulation (rTMS). For cases of treatment-resistant depression, deep brain stimulation has been successfully applied to the subgenual cingulate cortex (
18,
19) and the ventral striatum/nucleus accumbens (
20–22) without known permanent adverse effects in the subjects studied to date. Refinements in stimulation variables for rTMS applied to the dorsolateral pre-frontal cortex have significantly improved the magnitude and endurance of observed antidepressant effects (
23). While these techniques are safer than earlier rudimentary approaches to “psychosurgery,” the precise mechanisms by which deep brain stimulation and rTMS act are still incompletely understood. It is not known, for example, whether the local effects of deep brain stimulation work through excitation or inhibition or effects on fibers of passage (
24). Recently developed optogenetic tools make it possible to activate or inhibit particular neuronal cell types and/or their terminals within defined brain regions (
25), allowing for a deeper exploration of the neurophysiological mechanisms underlying the therapeutic effects of deep brain stimulation. Thus, as deep brain stimulation and rTMS are scaled down and characterized in laboratory animals, one can expect clinical improvements in patient selection, technique, and localization.
With heritability estimates of approximately 40% (
26), two main techniques have been utilized to explore the genetics of depression. Candidate genes, identified through an investigator's best guess about etiological mechanisms, have been examined through linkage and genetic association studies (
27). Single nucleotide polymorphisms (SNPs) in specific genes, such as GNB3 (for guanine nucleotide-binding protein 3) or MTHFR (for methylene tetrahydrofolate reductase), have survived stringent statistical requirements of meta-analyses. While their odds ratios are too weak for diagnostics or risk stratification, these and other genes may offer new clues into disease pathophysiology (
28). Genome-wide association studies are inherently unbiased, as they currently can simultaneously examine up to one million SNPs. While such trials have identified SNPs in previously unappreciated molecules, such as Piccolo (a presynaptic nerve terminal protein) or GRM7 (metabotropic glutamate receptor 7), these findings are themselves of relatively poor statistical significance and are not replicated across studies (
27). Several explanations have been put forth, including vague DSM diagnostic criteria, considerable disease heterogeneity, and the relatively potent contribution of ongoing life stressors and epigenetic plasticity. We remain hopeful that within the coming decade, that newer technologies will have greater success and replicability, including “whole-exome” studies (which exclude noncoding regions, representing 99% of the genome) as well as whole-genome sequencing. Of course, a key unanswered question is whether these genetic data should be correlated with DSM diagnostic categories, more broadly across several DSM diagnoses, or with more carefully defined behavioral, endocrine, neurochemical, or neuro-imaging phenotypes. Identifying such genes will be hugely beneficial for the generation of bona fide animal models of depression.
An important insight gained from everyday clinical practice is the observation that monoamine reup-take inhibitors and other modulators of monoaminergic function improve symptoms in about 50% of depressed patients and produce a remission in 30%–40% of patients (
29). These data illustrate the tremendous genetic heterogeneity of treatment response, and efforts are under way to identify pharmacogenetic predictors of a favorable treatment response to monoaminergic agents (
30,
31). Since monoamine enhancers improve depressive symptoms, it was suggested historically that depression is caused by deficits in monoaminergic transmission. This “monoamine hypothesis” continues to be a prominent preoccupation of the field. However, after more than a decade of PET studies (positioned aptly to quantitatively measure receptor and transporter numbers and occupancy) (
32), monoamine depletion studies (which transiently and experimentally reduce brain monoamine levels) (
33), and genetic association analyses examining polymorphisms in monoaminergic genes (
28,
34,
35), there is little evidence to implicate true deficits in serotonergic, noradrenergic, or dopaminergic neurotransmission in the pathophysiology of depression. This is not surprising, as there is no a priori reason that the mechanism of action of a treatment is the opposite of disease patho-physiology (
36). Thus, currently available agents likely restore mood by modulating distinct processes that are unrelated to the primary pathology of depression, just as diuretics improve the symptoms of congestive heart failure without affecting cardiac myocytes directly. Similarly, the success of intravenous ketamine in rapidly alleviating depressive symptoms in treatment-resistant depression (
37) has prompted an exploration of the cellular and neuroanatomical substrates for ketamine's actions and the search for ketamine-like therapies that lack psychotomimetic side effects. However, formulating a “glutamatergic hypothesis of depression” is grossly simplistic and only fuels inaccurate public misconceptions of depression's “chemical imbalance,” particularly since more than one-half of all neurons in the brain utilize glutamate as a neurotransmitter.
Animal Models of Depression
The design, application, and relative strengths and limitations of depression models have been discussed in several reviews (
1,
38). Without definitive knowledge of pathophysiological processes, these models are often evaluated for their
face, construct, and
pharmacological validity (
1), as are models of other clinically defined neuropsychiatric syndromes, such as autism and schizophrenia.
Face validity is a model's symptomatic homology to human depression. Today's depression models achieve this goal to a considerable extent: rodent and primate models have successfully recapitulated states of social withdrawal, hypophagia and weight loss, anhedonia, circadian changes, and abnormalities of the HPA axis, although these phenotypes are generally transient and not all present simultaneously.
The more challenging
construct validity is the ability of a model to replicate etiological factors implicated in depression, which are themselves not entirely understood. Most paradigms use some form of stress (of a physical or a psychosocial form), given the known association between independent stressful life events and depressive episodes (
39). More recently, a greater emphasis has been placed on replicating both environmental risk factors (such as stressful life events) and genetic risk factors (although these remain largely unknown) in the same model.
Pharmacological or
predictive validity is met when a model's depression-like behaviors are reversed by currently available antidepressant modalities, and several models in use today display this type of predictability with the therapeutic delay that characterizes antidepressant responses in humans. However, given that all available pharmacological agents are monoamine modulators and only a minority of patients experience remission after first-line therapies (
29), the requirement for pharmacological reversibility is perhaps desirable but not mandatory. Since the mechanisms underlying the delayed antidepressant effects of medication and nonmedication treatments (exercise, electroconvulsive seizures, etc.) remain largely unknown, animal models have been employed to dissect these mechanisms (i.e., models of antidepressant action), with the caveat that these therapies are applied to laboratory animals that generally lack depression-like behavior or any particular genetic vulnerability to depression.
A potential fourth criterion that has received considerably less attention is
pathological validity, whereby depression-related physiological, molecular, and cellular abnormalities in animals are validated by demonstrating identical changes in postmortem brain samples from depressed humans. This is a genuinely difficult requirement but has been gaining increasing popularity with the more widespread access to postmortem samples (
12–15). Ideally, this criterion might be better addressed through functional imaging studies with depressed patients, but this will require substantial improvements in molecular imaging capabilities.
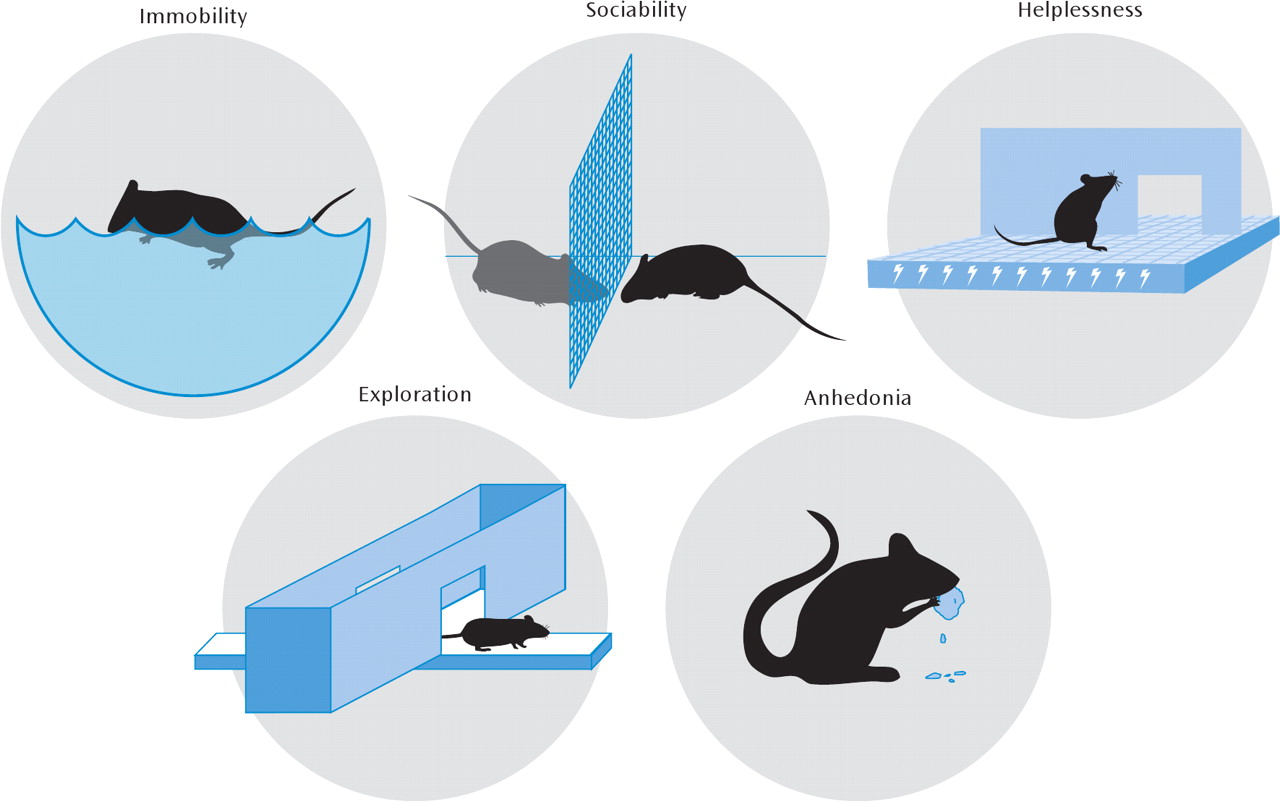
From an evolutionary perspective, depression may be an analogue of the “involuntary defeat strategy,” occurring when an animal perceives defeat in a hierarchical struggle for resources (
40). Hyperarousal, psychomotor retardation, reduced motivation, and sleep alterations in the setting of losing are postulated to have an adaptive advantage in that they serve to protect losers from further attack and focus cognitive resources on planning ways out of complex social problems (
41,
42). Most behavioral endpoints in depression models aim to quantitatively assay some type of experimentally induced defeat or despair (
Figure 2), even though this aspect of mammalian behavior is likely physiological (i.e., adaptive) rather than pathological. Additionally, while despair behavior is often extrapolated as being depression-like, it is clearly a huge inference to make from rodent models, and most stressors also produce anxiety-like changes that are exaggerated manifestations of the fight-or-fight response (reduced exploration, freezing, hyperthermia, HPA axis activation, etc.). For example, repeated social subordination in mice (social defeat) leads to a long-lasting phenotype of reduced social interaction with other mice. This impairment in sociability can be interpreted as a reduced motivation to interact (an abnormality of reward) or as a heightened avoidance of novel social stimuli (a pathological anxiety response). Distinguishing between these alternative hypotheses is difficult and may even be irrelevant, particularly given the poorly defined neurobiological distinctions between anxiety and depression and their highly variable clinical presentation. In either case, the model employs a naturalistic social-stress-induced behavior that is quantifiable and amenable to experimental manipulation (
12–14,
43–52).
The forced-swim and tail-suspension tests are the simplest and most widely used models of depression and antidepressant action. While these approaches have been rightly criticized for involving acute stress and acute antidepressant responses, they have permitted the rapid behavioral screening of novel chemical antidepressants and the phenotyping of genetically altered mutant mice. In certain instances, they have directed the field toward fundamentally novel molecular hypotheses. For example, an antidepressant-like phenotype in the forced-swim test (decreased immobility and greater struggling or swimming) was observed in mice deficient in acid-sensing ion channel 1a (ASIC-1a), a pH-sensitive ion channel expressed in the brain (
53). Subsequent studies have shown that ASIC-1a expressed in the amygdala participates in eliciting a fear response to a variety of aversive cues culminating in acidemia (
53,
54), implicating inhibitors of ASIC-1a (a previously unappreciated target) as potential therapeutics against anxiety and depressive disorders. Analogous approaches have identified several other novel molecular targets, including p11 (a calcium-binding chaperone molecule that promotes serotonin signaling through the serotonin 1B receptor subtype [15]), TREK-1 (a distinct type of potassium channel that is enriched in depression-related limbic brain regions [55, 56]), ghrelin (a stomach-derived endocrine mediator of energy homeostasis [46]), and many others.
In the following sections, we focus on neurobiological themes that exhibit therapeutic promise. The two main values of using rats and mice to study depression are 1) the ability to describe and characterize neuroplasticity with exquisite spatial and temporal precision and 2) the opportunity to utilize molecular innovations to demonstrate the causative effects of those neuroplastic changes on assays of depression- and antidepressant-like behavior.
Neurogenic and Neurotrophic Theories
The first description of continually dividing neuronal progenitors in the adult mammalian brain offered the promise of solutions for a host of neurodegenerative disorders that so far lack definitive cures (
57). Exploring the physiologic role of endogenous neurogenesis, particularly that which occurs in the hippocampal dentate gyrus, has important relevance to the study of psychiatric disease (
58). The journey from a hippocampal stem cell in the subgranular zone to a mature dentate gyrus granule cell neuron with appropriate synaptic connections occurs in stages defined by specific cellular markers, with the rates of proliferation and survival modulated by numerous stimuli. Unpredictable stressors, glucocorticoids, drugs of abuse, and high-energy electromagnetic radiation negatively infiuence this process, while antidepressants, voluntary exercise, and environmental enrichment accelerate adult hippocampal neurogenesis (
59).
Laboratory rodents have been used extensively to explore the regulation of these new hippocampal neurons and their contribution to depression-related phenotypes. In models of antidepressant action, cranial irradiation (which severely impairs the mitotic potential of hippo-campal stem cells) and aging (another robust negative regulator of adult hippocampal neurogenesis) impair some but not all of the effects of monoamine reuptake inhibitors (
60–63), suggesting that these agents may function through neurogenesis-dependent and -independent processes (
64). Clearly, only the actions of antidepressants that involve hippocampal circuitry could be mediated through enhanced neurogenesis. Indeed, one study was able to demonstrate the antidepressant effect of a direct intracerebral infusion of bone-marrow-derived mesenchymal stem cells, which both themselves transform into neurons and generate diffusible permissive factors that accelerate endogenous neurogenesis (
65). These preliminary results support the idea that enhancing hippocampal neurogenesis (pharmacologically or by way of cellular transplantation) can serve to boost or augment the antidepressant response. At the same time, impairments in the rates of neurogenesis do not appear to be involved in the core features of depression. Following cranial irradiation, mice are unimpaired across several indices of depression-related behavior (
60,
62). Consistent with its proposed role in hippocampal-dependent learning (
57,
59), adult hippo-campal neurogenesis may play a pathological role in the establishment of aversive memories of traumatic stressors and the sequelae of posttraumatic stress (
66). As the field struggles to clarify the functional relevance of these new neurons, stress-induced reductions in hippocampal proliferation are best interpreted as a marker of hippocampal plasticity (which may be impaired in some types of depression).
Another widespread endpoint for assaying the effects of stress, antidepressants, and genetic manipulations is the measurement of levels of brain-derived neurotrophic factor (BDNF) in the hippocampus. This practice, stemming from the “neurotrophic hypothesis” of depression (
67), is based on three main observations: an impairment of hippocampal BDNF signaling produces certain depression-related behaviors and impairs the actions of antidepressants (
68–70), experimental increases in hippocampal BDNF levels produce antidepressant-like effects (
71–73), and hippocampal BDNF levels are low in postmortem samples from depressed humans (
6). BDNF is one of numerous growth factors that have been implicated in depression, including firoblast growth factor, vascular endothelial growth factor, and nonacronymic VGF (
1). Through modulation of their levels and downstream signaling, these growth factors appear to transduce stressors into lowered rates of adult hippocampal neurogenesis, atrophic changes, and impaired synaptic plasticity of hippocampal neurons, which might (in theory) explain the cognitive impairments and hippocampal atrophy seen in depression (
67).
Translating these BDNF findings may not be straightforward. Aside from the challenges associated with synthesizing a specific agonist of BDNF, enhancing BDNF function in the nucleus accumbens and amygdala can have detrimental effects on measures of anhedonia, anxiety, and social interaction in rodents (
1,
71). A naturally occurring SNP in BDNF (G196A, Val66Met) results in dramatic alterations in intracellular trafficking of BDNF and its activity-dependent release (
1). Meta-analyses show that while the Met allele marginally increases the risk for depression in men but not women, it is also associated with a better antidepressant response (
74,
75). A hippo-campus-specific increase in BDNF activity may improve certain cognitive symptoms of depression and facilitate hippocampal neurogenesis (
60). While we possess the technology to deliver specific genes into the human brain through viral vectors (
76), the beneficial effects of BDNF would have to outweigh potential negative effects, i.e., lower seizure threshold, altered indices of learning and memory, and increased likelihood of malignant transformation (
77,
78). Nevertheless, understanding the roles of these growth factors in depression's pathophysiology remains an extremely active area of research, with an emphasis now placed on extrahippocampal trophic signaling and exploring downstream signaling pathways (
79), which may have greater pharmaceutical application.
Contribution of Epigenetic Modifications
Biological theories of depression's etiology have traditionally focused on the interplay between genetic risks and environmental/social hazards, with gene-environment interactions invoked to explain how relatively weak genetic vulnerabilities combined with the right environmental triggers may lead to significant psychiatric impairment (
80). However, the significant discordance of depression between monozygotic twins (who often share the same environment as well as genes), the remarkably slow progress in identifying genetic risk factors, and depression's twofold female predominance suggest the presence of a third, nongenetic and nonenvironmental component to variability (
81). Epi-genetic modifications have been implicated as a significant contributor to this third source of variability and are broadly divided into those that modify DNA directly (e.g., DNA methylation), those that alter histones (e.g., histone acetylation or methylation), and those that involve noncoding RNAs (such as microRNAs) that regulate gene expression (
82). In changing DNA's tertiary structure, they adjust interactions between DNA and associated proteins such as transcription factors and RNA polymerases, thereby ultimately altering levels of mRNA expressed by given genes. Pathological epigenetic events have been implicated in numerous chronic diseases, most notably cancer, in which aberrant epigenetic changes promote genetic instability (
83).
Through combining animal models with an explosion of novel molecular tools, several epigenetic events have been linked to depression-related behavior and antidepressant action. In rats, offspring born to mothers that display low levels of maternal licking and grooming behavior display exaggerated corticosteroid responses to stress and increased anxiety, which are mediated in part by increased methylation (and subsequent repression) of the glucocorticoid receptor gene promoter in the hippocampus. This type of epigenetic mark is stable to adulthood, reversed by chemical inhibitors of DNA methylation, and entirely dependent on the maternal behavior of the fostering, rather than biological, mother (i.e., independent of germ-line transmission) (
84). Early life stress applied to mice produces hypomethylation of the arginine vasopressin (AVP) gene in the hypothalamic paraventricular nucleus, resulting in hypersecretion of AVP, pathologically enhanced serum corticosterone level, and increased depression-like behavior (
85). Histone acetylation, a mark of active transcription, is increased at certain BDNF promoters when socially defeated mice receive a course of chronic imipramine, and this hyperacetylation event is mediated by the down-regulation of histone deacetylase 5 (HDAC5) (
47). While overexpression of HDAC5 in the hippocampus counteracts the effects of antidepressants, mice that are globally deficient in HDAC5 display an enhanced vulnerability to chronic stress (
49).
These examples illustrate the complexity in translating these epigenetic changes into clinical phenomena: while certain perturbations robustly alter epigenetic marks on one gene in one brain region, other brain regions may have opposing changes at distinct genes. Furthermore, most enzymes affected by epigenetic changes occur in several isoforms, each with its own tissue specificity and regulatory factors (e.g., HDAC5 is part of a family of 11 HDAC isoforms that are expressed across all major organ systems [
86]), further complicating the development of selective small-molecule antagonists. In spite of this complexity, epigenetic modulators show some promise as treatments for depression. In animal models, systemically or locally administered HDAC inhibitors display antidepressant properties without obvious adverse effects on health (
12,
82), suggesting that HDAC inhibitors may function by modulating a global acetylation/deacetylation balance across several brain regions. Of course, histone acetylation functions in concert with several other markers of gene repression and activation, including histone methylation, phosphorylation, sumoylation, and ubiquitination (
86). Thus, to comprehensively describe and appreciate the intricacies of depression-related epigenetic plasticity, we can expect a continued evolution in molecular and bioinformatic techniques. Rather than examining candidate genes such as those for BDNF and glucocorticoid receptors, the field has begun to transition toward genome-wide approaches to studying chromatin regulation (
48), shifting the focus from “epigenetic marks” to “epigenomic signatures.” As these technologies characterized in mouse and rat models begin to be applied to human postmortem tissue from depressed individuals (
87), the ultimate goal would be to use transcriptional and epigenetic profiling as biomarkers to distinguish clinical categories of depressive illness, to determine responsivity to various antidepressant classes, and to differentiate treatment-sensitive from treatment-resistant illness. These profiles may offer new insights into subtype-specific pathophysiology and therapies and aid in the validation of our current animal models.
Role of Dopaminergic Reward Circuits
The dramatic reinforcing properties of direct intracranial self-stimulation in rodents led to the appreciation of a series of subcortical regions critical for reward and appetitive behavior (
88). The two main structures implicated by intracranial self-stimulation are the lateral hypothalamus and medial forebrain bundle, the latter containing ascending dopaminergic projections from the ventral tegmental area to the nucleus accumbens (
88). Under baseline conditions, dopaminergic neurons in the ventral tegmental area oscillate between tonic patterns of activity (low-frequency regular action potentials) and phasic activity patterns (bursts of action potentials) (
89). Unexpected rewards produce a transient increase in phasic firing (encoding a “reward prediction error”), which is sufficient to reinforce antecedent behaviors (
25). All major classes of abused drugs appear to “signal” a reward, at least in part, by artificially enhancing dopamine transmission in the nucleus accumbens (for example, cocaine blocks the dopamine transporter) (
88).
Given depression's prominent features of anhedonia and appetite alterations, this circuit has become an obvious focus of attention for basic molecular and electrophysiological studies. In rodents, long-term antidepressant administration reduces the firing rates of dopamine neurons in the ventral tegmental area (
90). In contrast, psychosocial stressors activate firing in the ventral teg-mental area and increase nucleus accumbens dopamine levels (
13,
50,
91), and this may represent a positive coping strategy to enhance motivation during stressful situations (
88). One mechanism for this enhanced excitability of the ventral tegmental area may be the reduced activation of the protein kinase AKT, which leads to reductions in local inhibitory neurotransmission (
14). Variations in the neuroplastic adaptations expressed by these neurons may also contribute to individual differences in the responsiveness to stress. In the mouse social defeat paradigm, while stress-susceptible mice display enhanced activity in the ventral tegmental area and subsequent BDNF release, stress-resilient mice overcome this excitability change by up-regulating potassium channel subunits expressed by dopamine neurons in the ventral tegmental area that maintain normal tonic firing rates (
13,
44). Stress-induced increases in nucleus accumbens BDNF may mediate pathological reward learning such that, following a series of aversive social encounters, the positive rewarding value of social interaction is now modified to have a negative valence (
1). Enhanced mesolimbic dopaminergic signaling may explain the reported efficacy of antidopaminergic agents as adjunct antidepressants (
92), and by enhancing basal dopaminergic and BDNF signaling, this model may also explain the significant comorbidity of substance dependence and depressive disorders (
93,
94).
Nucleus accumbens neurons, anatomically situated to integrate reward-related dopaminergic signals as well as glutamatergic input from the prefrontal cortex, hippo-campus, and amygdala (
21), themselves display numerous stress- and antidepressant-induced changes (
88). One example is the modulation of cAMP response element binding protein (CREB): while prolonged social isolation stress reduces CREB activity and generates a predominantly anxious phenotype (
95), active stressors or drugs of abuse
increase CREB activity and promote anhedonia in the presence of a host of natural and drug rewards (
88). Neuroimaging studies with depressed humans show that quantitative indices of anhedonia are associated with low nucleus accumbens volume (
96) as well as hypoactivation during simple tasks of incentive reward (
97,
98). In an attempt to reverse this nucleus accumbens hypoactivation, bilateral deep brain stimulation to this and nearby regions has been successfully applied to several cases of treatment-resistant depression (
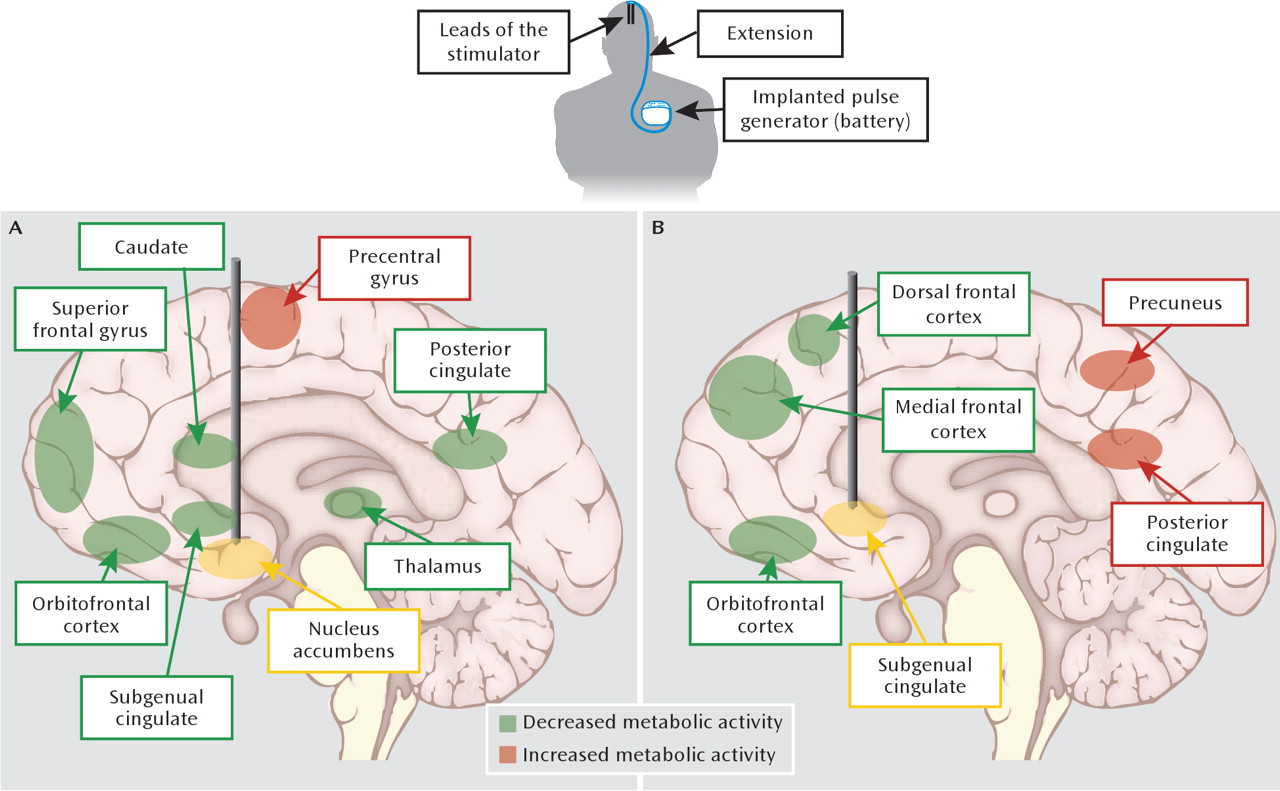
Figure 3). Consistent with the centralized location of the stimulation, responders displayed normalized PET indices of activity in the nucleus accumbens and the larger ventral striatum, in addition to lower activity in the subgenual cingulate cortex and other prefrontal cortical regions (
20,
21). In rats, deep brain stimulation applied to the nucleus accumbens with simultaneous electrophysiological recordings from multiple distant sites has suggested that the therapeutically relevant effects are due to the synchronization of inhibition across a network of cortical and subcortical regions (
99), possibly explaining anatomically distant effects of deep brain stimulation. In this way, the application and validation of deep brain stimulation in depression models offers opportunities to improve our circuit models (
Figure 1) and shed light on the neurobiological correlates of treatment resistance.
Sex, Steroids, and Immunity
The network of neural substrates involved in depression's symptoms displays a remarkable degree of plasticity in response to a host of peripherally derived chemical stimuli, and advancing our understanding of the endocrinology and immunology of depression offers exciting therapeutic avenues. Considerable research in the fild has focused on a central role of a pathologically dysregulated HPA axis (
1), whereby stress-induced hypercortisolemia leads to the central down-regulation of glucocorticoid receptors, impairing cortisol's negative feedback and enhancing levels of corticotropin-releasing hormone (CRH) and adrenocorticotrophic hormone (ACTH) (
36). This vicious cycle sustains elevated cortisol levels, possibly leading to hippocampal atrophy and reduced rates of neurogenesis, as well as predisposing depressed individuals to insulin resistance and abdominal obesity (
100,
101). A large body of clinical and preclinical evidence supports this model. Depressed patients display dexamethasone nonsuppression that is reversed by antidepressant treatment (
102), enhanced CSF levels of CRH (
103), and alterations in diurnal cortisol rhythms (
104). Mice that are treated chronically with glucocorticoids develop anhedonia in conjunction with other molecular correlates of depression (
105). In line with these data, chronic glucocorticoid administration reduces hippocampal volume and impairs cognition in humans (
106), while the glucocorticoid receptor antagonist mifepristone improves psychotic and depressive symptoms in patients with psychotic major depression (
107). Antagonizing CRH signaling, particularly through the CRH1 receptor subtype, leads to strong anxiolytic effects in several rodent models (
108). While the validation of CRH1 antagonists for depression and anxiety disorders remains an active area of clinical research, previously tested pharmacological prototypes have failed for a variety of reasons, including off-target hepatotoxicity (
109,
110).
This “cortisol hypothesis” represents a vibrant part of the preclinical depression literature: with commercially available glucocorticoid immunoassays, experimental manipulations are often validated as being “prodepressant” or “antidepressant” depending on their effects on baseline or stress-induced glucocorticoid levels. However, several key points argue for a reappraisal of this practice: 1) true hypercortisolemia is rarely observed in outpatient depressed populations and may be associated only with depression severe enough to require hospitalization (
111,
112); 2) depressed patients with atypical features and victims of posttraumatic stress tend to display hypocortisolemia (
111,
113,
114); and 3) mice designed to display reduced central glucocorticoid receptor signaling (mimicking hypercortisolemic states) and those that centrally overexpress glucocorticoid receptors display identical behavioral and endocrinological phenotypes (
115,
116). In spite of the strong immunosuppressant properties of glucocorticoids, levels of circulating proinfiammatory cytokines (taken as a quantitative marker of systemic glucocorticoid-receptor-mediated signaling) are usually elevated in major depression (
117); these cytokines include interleukin 1 (IL-1), IL-6, and tumor necrosis factor α. They are themselves sufficient to impair glucocorticoid receptor signaling, and thus, rather than directly affecting HPA function, stress likely leads to glucocorticoid
insufficiency through cytokine intermediates (
102). Under certain circumstances, this reduced glucocorticoid-receptor-mediated signaling may promote hypercortisolemia, severe insomnia, and hypophagia (melancholic features) but in other conditions may lead to hypocortisolemia, hyperphagia, and fatigue (atypical features). Cytokines themselves play powerful roles in depression-related neuroplasticity: chronic stress produces significant changes in immune function (
118), and cytokines induce depression-like behavior when injected into rodents (
119). IL-1β is one such cytokine: through the actions of the transcription factor nuclear factor κB, stress-induced increases in IL-1β lead to reductions in hippocampal neurogenesis and anhedonic phenotypes (
120).
The greater female predisposition to depression, as well as its greater incidence in postpartum and perimenopausal periods, argue strongly for a thorough understanding of the role of gonadal hormones in affective regulation. The heightened female vulnerability to experience depressive episodes is limited to the postpubertal and premenopausal period, and accordingly, much of the field's emphasis has focused on the neurobiology of estrogen. Studies in rodent models have demonstrated that estrogen has antidepressant properties and also augments antidepressant actions of monoaminergic agents. Conversely, mice lacking aromatase (required for the generation of estrogenic steroids) or estrogen receptor β display aberrant stress-related behavior (
121). Consistent with the broad central expression of estrogen receptor β, the antidepressant effects of estrogen signaling have been linked to several neurobiological substrates, including hippocampal neurogenesis, BDNF signaling, serotonergic neurotransmission, and HPA axis function (
122).
While this body of evidence may explain how significant fluctuations in hormone levels can trigger depressive episodes, it does not account for the heightened female vulnerability to depression, which is likely as much about female vulnerability factors in responses to depressogenic stimuli as about male resiliency factors. For instance, in comparison to males, female rodents display passive coping strategies and a more pronounced HPA axis activation in response to a variety of stressors. These features can be “masculinized” by providing testosterone during puberty, demonstrating how gender differences in behavioral physiology can be hardwired during certain critical periods (
123). Ovariectomy also promotes active stress-related coping, an effect that may be related to estrogen signaling within the nucleus accumbens (
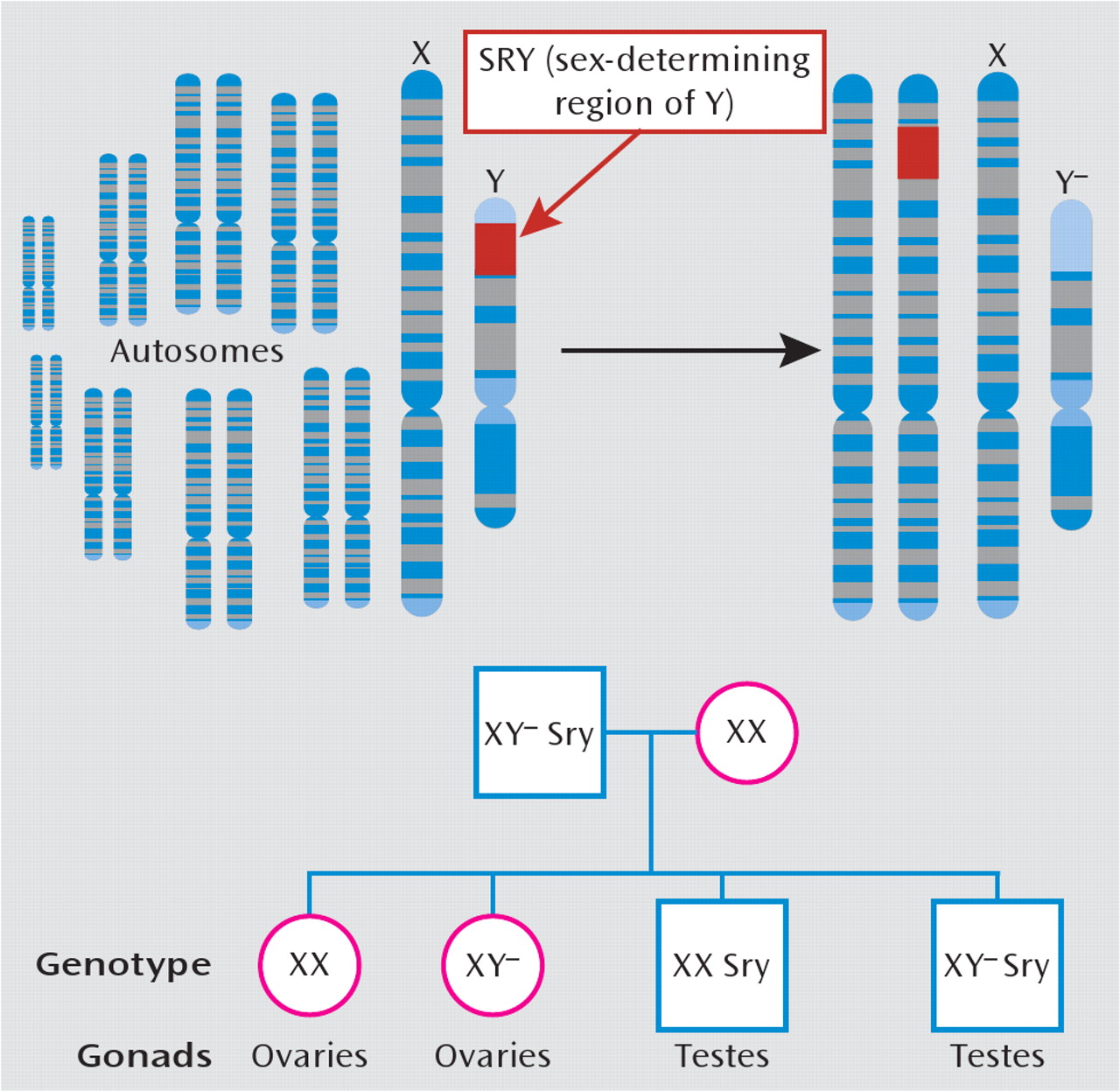
124). Aside from hormonal infiuences, it is important to recognize that gender differences also likely arise from numerous genes on sex chromosomes that are unrelated to gonadal function. Through standard genetic engineering techniques, one can create mice that are chromosomally male (i.e., XY) while having female gonads, and vice versa (
Figure 4). Studies with this model have shown that while the development and maturation of male copulatory behaviors and sexually dimorphic brain structures depend on gonadal output, other genes on sex chromosomes independently drive other behavioral traits that are relevant to depression, including habit formation, parental and aggressive behaviors, and social interaction (
125,
126).
Mediators of Energy Homeostasis
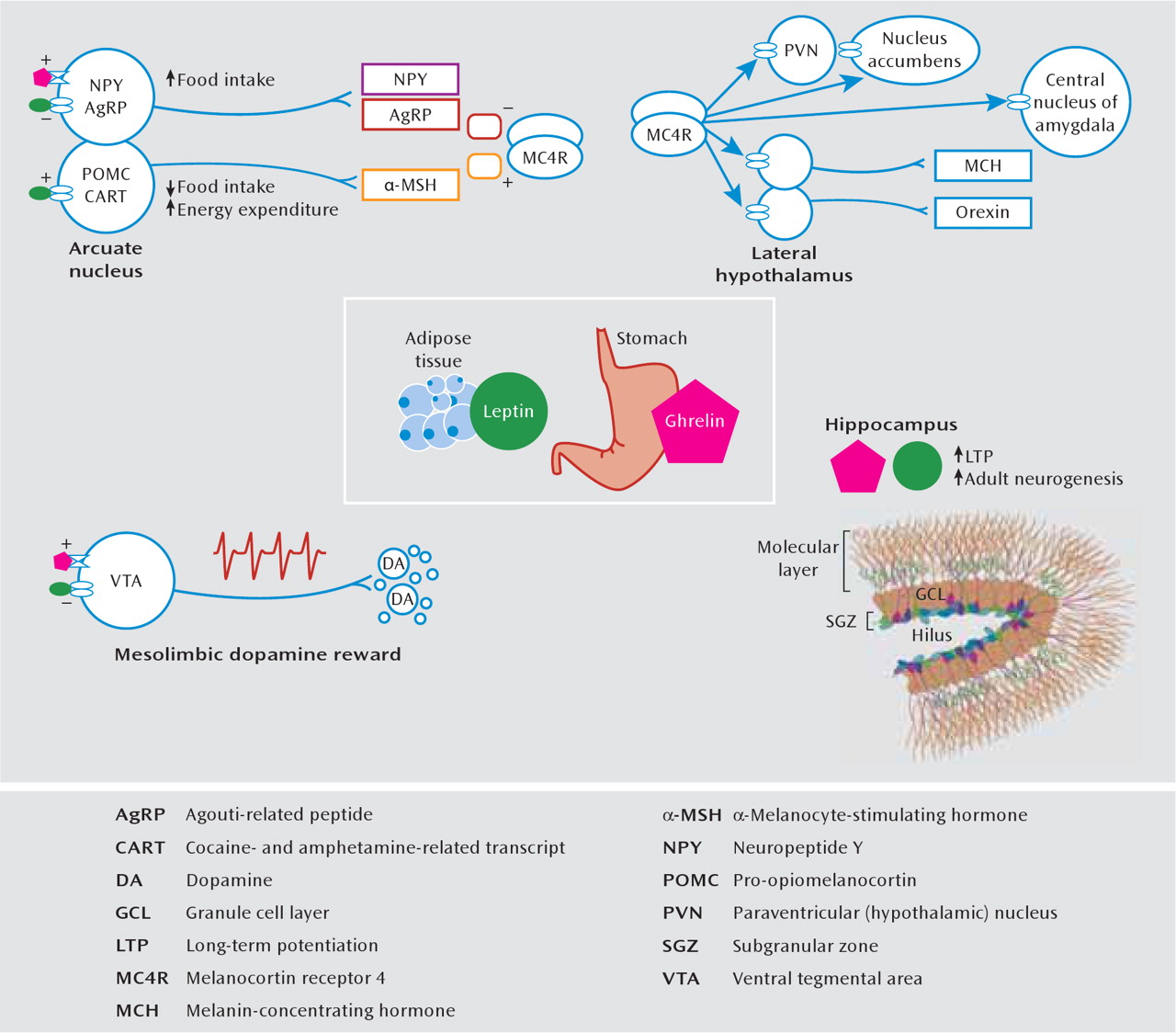
The appetite and metabolic abnormalities associated with depression and depression-related entities range from severe hypophagia and anorexia to binge eating and obesity. A thorough understanding of such complex phenomena requires knowledge about physiological mechanisms of energy homeostasis, which refers to processes that maintain equilibrium between caloric intake and energy expenditure. In mammals, this is achieved largely through the action of circulating hormones that relay information about peripheral energy levels to the brain (
127). Two such hormones that have received tremendous attention are leptin and ghrelin (
Figure 5). Leptin is synthesized in white adipose tissue and is secreted in times of nutritional excess. Many obese individuals display a hyperleptinemia associated with central leptin resistance (
131). In contrast, ghrelin is synthesized by gastric fundus cells and released during times of energy scarcity, and its secretion stimulates caloric intake and energy storage (
127). The principal homeostatic site of action of leptin and ghrelin is the hypothalamic arcuate nucleus, where they exert anorexigenic and orexigenic effects, respectively, through a biologically elegant system of neuropeptides. It is interesting that receptors for leptin and ghrelin and receptors for other feeding-related peptides (such as melanin-concentrating hormone, neuropeptide Y, agouti-related peptide, α-melanocyte-stimulating hormone, and orexin [hypo-cretin]) are expressed in several depression-related limbic substrates. In rodents, chronic stress decreases serum leptin levels (
132) and increases serum ghrelin (
46). The systemic administration of either hormone produces antidepressant effects on the forced-swim test, enhances hippocampal neurogenesis, and improves learning and memory in behavioral and cellular (i.e., long-term potentiation) assays (
46,
132–136). Whereas ghrelin and leptin have identical actions in the hippocampus, dopaminergic neurons of the ventral tegmental area are excited by ghrelin and inhibited by leptin (
130,
137,
138), which illustrates how their hypothalamic effects on appetite are complemented in the ventral tegmental area through opposite modulation of reward sensitivity.
In addition to persistent deficits in social interaction and anhedonia, mice subjected to chronic social defeat stress display an initial weight loss followed by a prolonged hyperphagic phase, during which they rapidly regain their body weight and eventually gain more weight than do control or stress-resilient animals. This phenomenon is at least partially mediated by both reduced serum leptin levels and central leptin resistance, which ultimately weaken central melanocortinergic signaling, i.e., through the melanocortin 4 receptor (
Figure 5). This hypoleptinemia seems to be mediated by enhanced β
3-adrenergic signaling, which promotes sympathetically mediated lipolysis. Coadministration of β
3-adrenergic antagonists during social defeat prevents the weight gain and reduced leptin but worsens social deficits (
51), suggesting that enhanced β
3-adrenergic signaling has an adaptive function at the expense of metabolic derangements.
Understanding the hedonic impact of homeostatic signals provides numerous targets for pharmaceutical development in depressive disorders, particularly in cases associated with significant metabolic abnormalities. An obvious example would be in cases of HIV- or cancer-related cachexia, where artificially enhancing ghrelin or attenuating melanocortin signaling would have therapeutic hyperphagic and antidepressant effects. Nonpeptide antagonists of the melanocortin 4 receptor have already been shown to exert antidepressant and anxiolytic effects in animal models (
129). Conversely, patients with comorbid depression and obesity (
139) might benefit from therapies designed to alleviate central leptin resistance (a challenging objective). Elucidating such therapies will require a deep understanding of the anatomy and physiology of leptin receptor signaling and of numerous other feeding-related peptides. This is an exciting area of active research.
Conclusions and Perspectives on the Future
Today's approaches to dissecting the neurobiology of depression employ an unprecedented array of experimental techniques in humans and animals, including genome-wide DNA sequencing, chromatin immunoprecipitation to study epigenetic factors, functional brain imaging, opto-genetic electrophysiological tools, viral-mediated gene transfer, and an impressive assortment of genetic mutant mice. The list of molecular players involved in depression's phenotypes has now expanded to include genes from diverse aspects of cellular physiology, such as numerous neurotransmitter and neuropeptide systems, steroid hormones, neurotrophic and cytokine signaling cascades, ion channels, histone deacetylases, circadian genes (
88,
108), transcription factors (e.g., CREB, nuclear factor κB, and ΔFosB [
43]), p11, and many others. Most of the new targets are derived from experiments in rodents and while these studies are scientifically sound, the targets themselves may or may not be therapeutically relevant or feasible for human depression. Beyond the synthetic obstacles to designing safe and effective small-molecule modulators or viral vectors for use in depressed humans, a key challenge will be to prioritize these targets and develop collaborative efforts to rule them in or out at a reasonable pace.
A large and unacceptable divide continues to exist between animal studies and human clinical investigation. An important example involves our appreciation of cortical contributions to depression: while human neuroimaging studies repeatedly implicate cortical subregions, such as the subgenual cingulate and orbitofrontal cortex, the vast majority of rodent studies limit their analyses to the hippocampus or amygdala. While studying cortical circuits in rodents is more challenging, the human findings clearly demonstrate the high priority of this work. Many reports in the basic literature focus on drastic behavioral and neurobiological phenotypes in constitutive knockout mice, even though homozygous human “knockouts” presumably are a negligible contribution to clinical depression. As progress in delineating the genetics of depression continues, it will be crucial to complement knockout studies by examining molecular and epigenetic mechanisms underlying individual variability and understanding the cellular and physiological consequences of psychiatrically relevant human SNPs. Finally, pathological validation using postmortem brain tissue provides a crucial link between our inherently limited laboratory models and the molecular enigmas of human depression.
Human studies must also mature. Observational studies that measure serum BDNF or glucocorticoid levels can expand to include multiple measures such as serum leptin, ghrelin, and thyroid hormone levels and metabolic status, to name just a few, as well as segregating patients into depressive subtypes. Brain imaging experiments continue to largely focus on volume or activity measures of particular brain regions or on monoamine receptor/ transporter occupancy. It is essential to vastly expand the range of proteins that can be assessed in the living brain so that proteins at the heart of pathophysiological models in rodents can at last be analyzed in depressed patients. As informed clinicians and scientists in the field, we have a responsibility to expand the horizon of our investigations and constantly reassess our analytic methods and theoretical paradigms. We should look well beyond monoamines, cortisol, BDNF, and the hippocampus to determine tomorrow's novel medical and surgical therapeutic avenues for depression.